- +1
对抗“罕见病中的罕见病”,是一场延缓退化的赛跑
南京男孩满满(化名)的一天,是从过程艰辛的刷牙开始的。
为协助其完成这项任务,母亲张亚欢特意网购了一套牙科工具。每天早晚,她会先将咬合垫塞在满满的后牙根处,再用牙刷清洁其牙齿。清洁后产生的泡沫,他没法通过漱口吐出,张亚欢只能拿沾了温水的纱布一颗一颗牙齿地擦过去。
“你能联想到的吃饭、洗澡、大小便,(他)都需要人帮助,不能自己独立完成。”张亚欢在接受澎湃新闻(www.thepaper.cn)采访时说。
这些生活障碍,遍布满满的生活中,就像在其体内越积越多的黏多糖。这种长链复合糖分子用于构成人体骨骼、肌腱及其他组织器官,正常情况下,老化的黏多糖可以通过溶酶体中多种酶的催化被分解;但由于缺少α-N-乙酰葡糖苷酶,满满体内的黏多糖开始在溶酶体中贮存,随着时间的推移,它们将更强势地侵害他的器官,剥夺其正常生活的能力。
这种渐进式的侵害,对于满满及其家人,都是残酷的。
黏多糖贮积症患者的绝望,在于无法逆转的机能退化
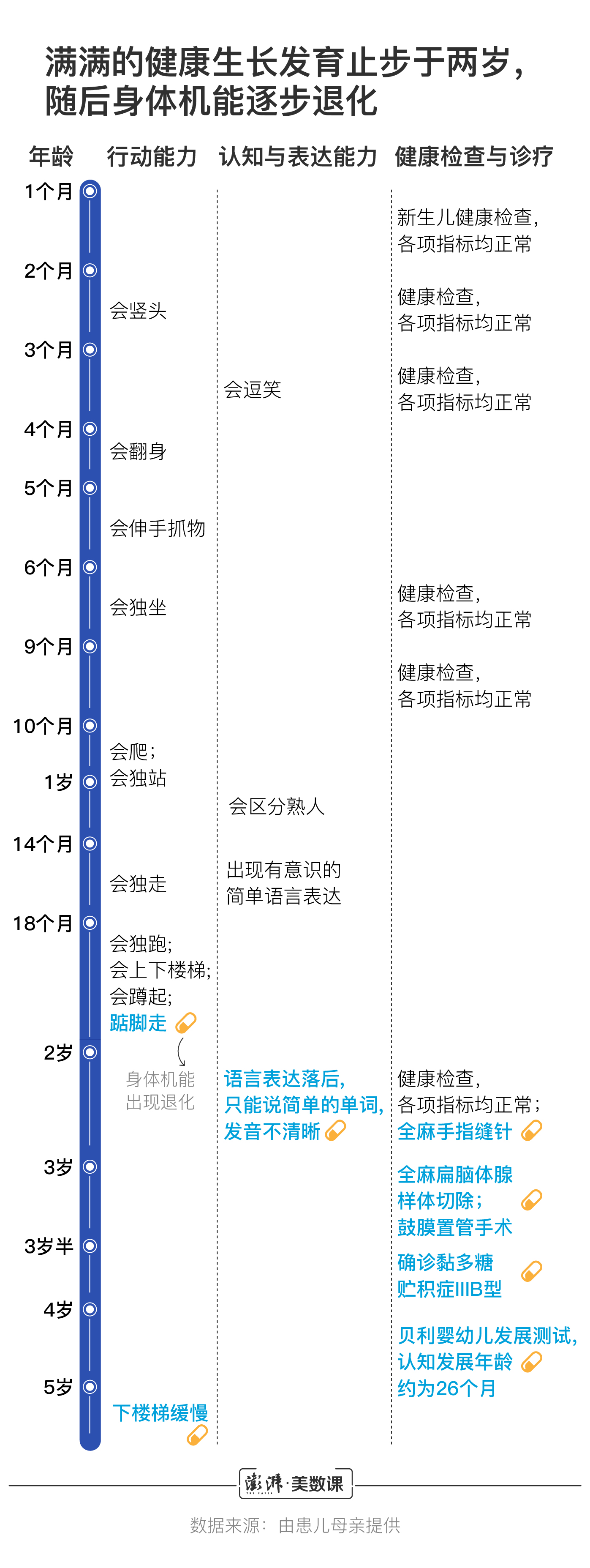
在三岁半被确诊为黏多糖贮积症IIIB型之前,满满曾拥有长达两年的健康生长记录。他从学会竖头开始,逐步掌握翻身、伸手抓物、独坐、爬、独走等技能。他不仅健康,而且聪明。14个月时,他开始学说话,不到两岁就可以清晰地哼唱儿歌,用简单的词句表达自己的需求。
和其他孩子一样,他积极地为成长成一名独立的个体做着准备,直到身体机能因黏多糖的过度堆积开始退化。
他总是表现得过分活泼,像一只脱缰的小野马。对他发号施令并不管用,外出时家人得尽力用手将其牢牢牵住,不然他很容易闯祸。
语言交流是极为困难的,他目前所能使用的都是一岁多时掌握的简单词汇。一天之中他最常念叨的是“爸爸”,但这并非出于对爸爸的想念和热爱,因为当爸爸真正出现在其面前时,他仍会“对着其他的地方喊爸爸”。
“他嘴里说的‘爸爸’的意思,其实是混乱的,他现在的语言表达能力很差,没有办法清楚地表达自己的意思……因为语言能力退化了,‘爸爸’比较好发音,他每天‘爸爸爸爸爸爸’要叫上几百次。”张亚欢在接受采访时说。
为了照顾满满,张亚欢辞去了工作。尽管承受的辛苦是“正常家庭很难想象的”,但她宁愿这段总是超乎其掌控的多动期能一直延续下去。
因为当病情继续发展,满满最终将“逐渐安静下来”。这是黏多糖贮积症IIIB型病程发展的最后一个阶段,张亚欢将面对的是一个看似可控但绝望的境地。像最初一步步丧失语言能力一样,满满的脚步会越来越不稳,最终将失去行走的技能,喜爱美食的他还将失去吞咽功能,必须依靠胃管吸收流食,智力会进一步退化,最终他将像阿尔茨海默症患者一样,无法辨认家人。
医生预估,满满的寿命只有十几年。
黏多糖贮积症是“罕见病中的罕见病”,确诊取决于“决心”
国际黏多糖组织将每年的5月15日定为国际黏多糖日,用以“赞扬和鼓励与黏多糖贮积症斗争的患者及其家人,怀念因此病而离世的孩子们,并感谢致力于研究和治疗黏多糖贮积症的医生和科学家们”。
在南京,满满一家或许是唯一纪念这个日子的IIIB型家庭。
张亚欢加入的IIIB型病友群,共有三十几位群友,他们“散落”在全国各处,彼此间的交流主要在线上,尚未有线下齐聚的机会。
人数稍多的是聚集了黏多糖贮积症各型病友的微信群,共有两三百人。北京正宇黏多糖罕见病关爱中心的创始人郑芋,是这个群的牵头人。她将黏多糖贮积症形容为“罕见病中的罕见病”,根据她的估计,中国的患者数可能在3000人左右。
这个数字,是郑芋根据黏多糖贮积症的患病率预估的。中国目前尚未有机构做过详细的患者统计,以欧美国家的统计数据来看,黏多糖贮积症不同型的人群比例差异很大,最常见的I型、II型,患病率大致为十万分之一,这个数值在较少见的III型中可以低至一百万分之一。
如果将这两个数值与欧美罕见病患病率的定义对比——美国和欧盟分别为6.37/10000和5/10000,郑芋对黏多糖贮积症罕见程度的形容无疑是准确的。
考虑到罕见病误诊率高的现状,郑芋认为中国黏多糖贮积症患者群体可能比3000人次更庞大。
黏多糖贮积症被归类为先天性遗传代谢病,成因是患者体内遗传基因细胞缺乏某种能分解黏多糖的酶,导致黏多糖在体内细胞中过量堆积,最终影响细胞的正常功能并损害各个器官。
尽管发病原理解释起来很简单,但临床表现却颇为复杂。根据不同患者所缺乏的酶的差异,黏多糖贮积症被分成I型、II型、III型、IV型、VI型、VII型和IX型这七大型,前四种又可以被细分成几种亚型。不同型之间、同一型但处于不同病程的患者之间,病理表现均存在差异。

在病痛挑战基金会、香港浸会大学和华中科技大学发布的《2018年中国罕见病调研报告》中,参与调查的黏多糖贮积症患者有109名,曾被误诊的有83名,是未曾被误诊者的3.2倍。
多数患者在幼儿期或儿童期就已发病,儿科是他们均会前往就医的科室之一。但根据欧美学者2016年时发布的调查数据显示,即便在北美和欧洲,对于黏多糖贮积症中患病率较高的I型,熟悉它的儿科医生数量也极少。

最初她以为是舌脐带过短导致的发音不清。在南京市口腔医院检查后,这一疑虑被否定了,医生认为问题可能出现在听力上。于是,她又带着孩子前往南京儿童医院耳鼻喉科就诊。听力的确出了问题,满满被鉴定为极重度耳聋,但耳蜗检查却显示一切正常,这意味着病因可能集中在中枢神经系统上。
“医生建议我做的所有检查都做了,基因检测我们家就做了三次。”张亚欢告诉澎湃新闻。
最后,经过口腔科、耳鼻喉科、神经内科、康复科、儿科,辗转南京、上海和北京的多所医院,耗时八个月,满满的病最终在北京协和医院得到确诊。
“我就一定要查出病因,有这种精神在,才能短时间确诊。正常如果你问群里的其他家长,基本上没有这么早确诊的。”张亚欢解释说。
《2018年中国罕见病调研报告》曾调查了近2000名罕见病患者,总结发现,“当年确诊率低、确诊需辗转多所医院及多个科室”,是常态(详见澎湃新闻《国际罕见病日|平均5年确诊背后,待解决的不只是医学难题》)。
确诊的效率,如张亚欢所说,很大程度上取决于患者家属查出病因的“决心”。
如何用上药,是确诊之后更具挑战的部分
黏多糖贮积症几乎不可能被治愈,但目前I型、II型、IV型、VI型和VII型均可通过酶替代疗法——一种向患者注射含有缺失酶药剂的治疗方法——延缓病情的加剧。
在女儿确诊四年后,郑芋了解到美国已经在研发相关的药物。2008年到2012年,她频繁地前往台湾,追着在当地推行试药实验的美国药厂,期待为女儿争取到用药的机会。
“那个时期就是追药的过程,因为有希望了,所以我们就要追着希望跑。药厂在哪里办活动,我们就带着孩子去哪里参与。”郑芋告诉澎湃新闻。
但她的试药申请最终被美国方面驳回,原因是中国大陆没有医保保障后续的用药,患者很可能因为没有足够的经济能力支持终身用药,而被迫断药。这种结局在他们看来是不人道的。
中国台湾地区的黏多糖协会会长告诉她,那里的患儿有试药的机会,是努力12年争取来的结果。“人家说你们大陆的孩子要用药,得靠你们自己。那个时候我就埋下了种子,我们必须抱团发声,我们得自己争取用药的机会,不能去求着别人。”郑芋说。
2012年,她创立了北京正宇黏多糖罕见病关爱中心,希望将散落的黏多糖贮积症家庭聚集在一起,通过各类宣传活动,让社会关注并了解这个群体。
2014年,通过一场慈善晚宴,关爱中心收获了70多万的筹款。郑芋拿着这些钱,却陷入了迷茫。她不知道应该如何使用这笔钱,因为如果要让黏多糖患者用上药,“一个人一年的治疗费用最少在100多万”。
“我当时内心很绝望,觉得这个路好遥远,即使是想帮助一个孩子都很难,杯水车薪的感觉,当时我们中心大概已经有100多个患者,如果要为这些孩子筹到一年的费用至少需要几千万。”郑芋回忆道。
在患者家庭的每日生活中,这种绝望是具象的。在一些病情严重的患儿身上,家长每天都能看到一些退化。
“家长希望早点看到有药,越是已经知道有药了,但是觉得我们还够不到,可能就越会有一种自责,就是为什么我的能力还是达不到。我觉得这是每一天很多家人都挺困扰的问题。”郑芋说。
直到近两年,情况才真正乐观起来。
2018年5月,国家卫健委、科技部、工业和信息化部、国家药品监督管理局、国家中医药管理局五部委联合发布了《第一批罕见病目录》(共收录121种罕见病),这被视为朝罕见病立法迈出的一大步。
2019年3月1日起,根据国务院常务会议的决定,对首批21种罕见病药品和4种原料药,进口罕见病药品减按3%征收进口环节增值税,国产药则选择按3%简易办法计征增值税。税价联动下,罕见病患者的医药负担可能会因此减轻。
郑芋认为,促成改变的原因和罕见病群体“形成团体的意识”有关,中国目前有7000多种罕见病,依次成立起来的罕见病组织从初期的十几家,在近几年发展到上百家:“大家在不同时间不同地方发出声音,让政府注意到了这些问题。”
对于黏多糖贮积症患者而言,更积极的改变出现在去年11月和今年3月。国家药监局药品审评中心发布了两批“境外已上市临床急需新药名单”,计划加快审批这些药物在国内的上市,以满足罕见病治疗的需求;其中包括四种黏多糖贮积症药物,分别对应四种病型。

要解决这个问题,郑芋和其他患者家庭需要作出新的努力——与地方医保机构商谈是否可以与药厂协商药价,最后设置适当的报销比例以减轻患者的经济负担。
“这是下一步的事情。但如果没有药,就没有下一步。即使地方医保说我们对罕见病有多么关注,有什么政策可以解决问题,报销多少比例,对于我们来说,就是空谈,没有药的啊。有药是医保替我们来支付的前提条件。”郑芋说。
但对于黏多糖贮积症III型患者,未来仍是一个未知数。这种疾病目前在全球范围内还没有上市的药物。由于病灶在中枢神经系统,这种病型的药物研发尤为困难,适用于其他型的酶替代疗法并不能在III型患者身上奏效,因为被注射入体内的酶无法通过血脑屏障进入大脑。
而推动药物研发,不是依靠抱团发声、呼吁社会关注就能实现的。
“罕见病,不是说你家卖房卖车就能解决的。”张亚欢说,“我觉得得靠政府、企业和社会各界的力量去推动。现在国内药企愿意去做研发的也比较少。”
因为“没有其他更好的选择”,张亚欢也曾申请过美国药厂的试药实验。最终被拒绝的原因是满满智力退化严重,他目前的智力水平已远低于实验标准。即便成功试药,满满的病情也无法逆转:“最多也就是延缓寿命,没办法让孩子以后像正常孩子那样学习生活。”
去国外参加黏多糖会议时,张亚欢曾见到过黏多糖贮积症其他病型的患者,一岁多就开始用药,病情控制良好,看上去和正常孩子无差。“他们一确诊马上就用药了,还是挺羡慕的。”张亚欢说。
她现在能为满满做的,是通过每周五天的特校学习、每周两个课时的语言康复训练和日常的基础护理,尽可能延缓病情的加重。
张亚欢期待药物的上市。“如果上市的话,即使要花很多钱去买,我也想买回来试一下。即便不能终身用,但是尽可能让他试试看,有这个酶是什么样的感觉。”顿了一下,她补充了一句:“应该要等好久。”



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

