- +1
【重磅综述】Cell重磅综述|全面解析衰老与癌症的关系
以下文章来源于老顽童说 ,作者老顽童说
老顽童说.
公众号致力于传播衰老相关的前沿科研进展和趣味科普,帮助大家更深入地了解衰老背后的科学故事~

关注我们,获取更多相关资讯
翻译 by 张保虎、左越晟、于妍、宁太鑫、王雪宝
如果说衰老是为人生画上句号而奏响的圆舞曲,癌症则更可能是一场草草收场的急奏曲。芸芸众生,谈癌色变,而谈到衰老更多的是无奈,也是对美好一生的留恋。哲学上认为,万事万物皆有联系,而衰老和癌症也不例外,且更显错综复杂。2023年1月3日,来自西班牙奥维耶多大学的Carlos López-Otín教授和法国古斯塔夫·鲁西研究所的Guido Kroemer教授等人在Cell Metabolism杂志上发表了题为“Meta-hallmarks of aging and cancer”的综述,全面总结了衰老和癌症之间的或相同、或促进,或拮抗的错综关系。
摘 要
衰老和癌症具有一些部分重叠的“特征”。基因组不稳定性、表观遗传改变、慢性炎症和生态失调,这些衰老的特征与特定的癌症特征十分相似,因此构成了共同的“元特征”;而端粒缩短和干细胞耗竭等衰老特征会抑制肿瘤的发生,因此可以视为主要的“拮抗特征”。巨自噬失能和细胞衰老是衰老的两个特征,发挥着环境依赖性的肿瘤抑制和肿瘤促进作用。同样,与衰老相关的营养感应失调和与癌症相关的细胞代谢变化之间的等效或拮抗作用亦是复杂的。如何更好的理解癌症发病率和死亡率与年龄的正相关性和在更长寿人群中的负相关性,需要更深入的了解驱动衰老和癌症过程之间的促进性和拮抗性关系。同时,这种复杂关系的明晰也能为老年人恶性疾病治疗策略提供参考。
介 绍
无人能置衰老于度外,这表现为维持年轻与健康的整个(生态)系统的逐渐解体崩坏。然而,癌症只在特定阶段影响约三分之一的女性和一半的男性,这表明恶性疾病的发生并非不可避免——患病的“厄运”可能只会降临于我们中的一部分人,而健康的生活方式和有利的基因可以保护我们免受疾病的侵害。然而,衰老仍然是各种癌症的最重要风险因素,癌症发病率随着年龄的增长而上升,在85岁时达到峰值。有趣的是,90岁以后,癌症发病率和死亡率呈现净下降的趋势,100岁以上的癌症发病率和死亡率下降到5%以下,而呼吸系统/传染病和神经退行性疾病的死亡人数却急剧增加。衰老和癌症发生的这种双向关联性可以被解释为某些衰老的机制刺激癌症的发生和发展,而其他衰老相关机制则限制了癌症的发展。
值得一提的是,与未罹患癌症的老年人相比,超过65周岁的罹患癌症的老年人,其合并症以及衰老相关疾病的发病率增加。这一关联性可以用两个互不排斥的假设来阐释。一种假设是生物衰老的晚期阶段(合并症可被视为这一阶段的生物标志物)可能会诱发癌症。相对地,另一种假设是由于癌症对其他器官及肠道微生物群的远距离影响,癌症的发展可能会加速机体健康状况的恶化。此外,癌症治疗无论是局部治疗(手术或放疗)还是全身治疗(化疗、免疫治疗等),都会诱发机体压力响应和全身炎症反应,从而加速衰老进程。因此,衰老与癌症之间的关系是双向的。常见的生活方式因素(如吸烟和肥胖)的存在进一步加强了这种关系的复杂性,这些因素既增加了衰老的速度,也增加了患癌的风险。此外,一些遗传综合征表明衰老和癌症存在共同的遗传基础,即衰老加速(早衰)可能伴随着多种癌症的早发。
在此,作者对衰老和癌症的特征进行了系统解析。Cell期刊于2000年由Hanahan和Weinberg发表了“癌症的特征”的综述文章,并于2011年发表了文章的新版本,Hanahan最近在2022年的Cancer Discovery期刊中更新了这些特征。同时,Cell期刊于2013年和2023年由López-Otín等人发表了“衰老的特征”的综述文章。所有这些论文都指向同一结论,即从整体上看,无论是癌症还是衰老都无法用一种单一的分子通路来解释。相对地,癌症和衰老的显著特征可以通过以下三个标准进行列举:(1)其在所研究的现象之前出现或伴随所研究的现象;(2)其在实验进程中引发癌症或加速衰老;(3)其实验性或治疗性的靶向作用在模式生物中可以抑制癌症发生和发展,或延缓甚至逆转衰老。根据目前已发表的文献,提出了12个衰老特征及14个癌症特征。衰老的特征包括基因组不稳定性、端粒缩短、表观遗传改变、蛋白稳态丧失、巨自噬失能、营养感应失调、线粒体功能障碍、细胞衰老、干细胞耗竭、细胞间通讯改变、慢性炎症和生态失调。癌症的特征则包括维持增殖信号、逃避生长抑制、抵抗细胞死亡、无限复制的能力、促血管生成、激活浸润和转移、基因组不稳定性、炎症反应、能量代谢重编程、逃避免疫清除、解锁表型可塑性、非突变表观遗传重编程、衰老细胞以及多态的微生物组。
总的来说,衰老和癌症的许多特征非常相似,表现出很强的等效性(图1)。在此,作者将这些共同特征(基因组不稳定性、表观遗传改变、慢性炎症和生态失调)称为衰老和癌症的元特征。衰老的一些特征(蛋白稳态丧失、线粒体功能障碍和细胞间通讯改变)在癌症中缺乏明确的对应特征,但其可能对癌症的特定特征起诱导作用,有利于癌症中的细胞程序性死亡抵抗、糖酵解代谢和组织结构破坏。衰老的其他特征(端粒缩短和干细胞耗竭)则抑制了某些癌症特征的出现(无限复制的能力和表型可塑性),作者将这些特征定义为拮抗性特征。衰老的另两个特征,巨自噬失能和细胞衰老,它们具有环境依赖性的抑癌作用和促癌作用,因此需要单独的讨论。此外,衰老相关的营养感应失调与癌症相关的细胞代谢紊乱之间的等效性和拮抗性是极其复杂的,需要对衰老和癌症的常见代谢特征进行检查后研判,而癌症的逃避免疫清除这一特征则可能会受到几个衰老特征的共同影响。文章的最后,作者讨论了年轻和老年癌症患者抗癌治疗应答有何不同之处。
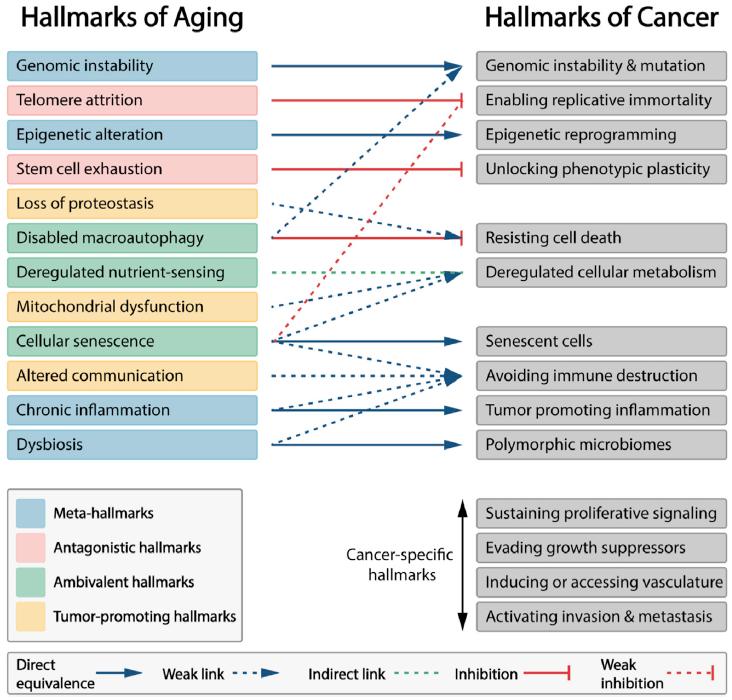
图1. 衰老和癌症的特征概述。
箭头说明了衰老特征(左)和癌症特征(右)之间的关系。依据衰老特征与癌症特征间的不同关系,衰老特征在图中被标注了不同颜色,并可归类于等效性特征(元特征)、拮抗性特征、矛盾性特征和促癌特征。在这些不同类别的衰老特征中,元特征和促癌特征有助于癌症的发生和发展,拮抗性特征则主要起防止或减缓癌症发展的作用,矛盾性特征可以促进或抑制癌症的进程。
具有致癌作用的衰老特征
4种衰老特征(基因组不稳定性、表观遗传改变、慢性炎症和生态失调)与4种癌症特征 (基因组不稳定性、非突变表观遗传重编程、炎症反应和多态的微生物组)有密切的相似性。因此,作者将其列为代表衰老和癌症的元特征(图2)。
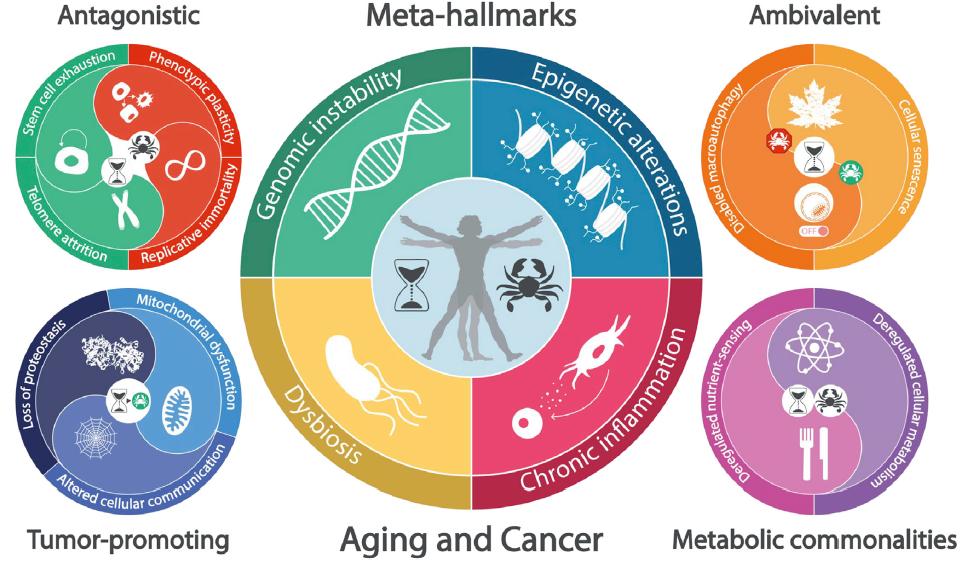
图2. 衰老和癌症的元特征。
中心环总结了本文提出的衰老和癌症的四个元特征:基因组不稳定性、表观遗传改变、慢性炎症和生态失调。侧环则分别对应拮抗性特征(端粒缩短和干细胞耗竭)、矛盾性特征(巨自噬失能和细胞衰老)、促癌特征(蛋白稳态丧失、线粒体功能障碍和细胞间通讯改变)和同时影响衰老和癌症的代谢改变(营养感应失调和细胞代谢失调)。
基因组不稳定性
生物体基因组的完整性受多种环境及内源因素影响,这将引起各种分子水平上的改变,并最终导致衰老和癌症。在衰老过程中,人类和模式生物的核DNA积累了体细胞突变、基因拷贝数变异和染色体非整倍体,这些突变和变异可能影响具有关键生物学功能的基因,并产生细胞突变、组织畸形和器官缺陷,进而导致衰老及与衰老相关的病理反应。在癌症进程中,染色体水平的不稳定性会引起染色体大范围区域或核苷酸水平的丢失和增加,导致碱基改变或核苷酸的插入和缺失,最终导致癌细胞的恶性转化。针对基因组不稳定性,所有物种都进化出了一套复杂的DNA修复系统,以尽量减少不可避免的遗传损伤并维持细胞稳态。然而,这些修复机制并不能修复所有的DNA损伤,且随着个体衰老逐渐失去效率。细胞中基因组不可逆损伤的累积将会导致生物体对癌症和其他衰老相关疾病的敏感性增加。正常的人体组织在个体年轻时每个细胞已表现出数百种突变,而随着个体的衰老,这一数字将会达到一个更高的数量级。同样,泛癌基因组研究表明,癌细胞积累了大量突变,根据其对癌症发展和进程的影响,这些突变可被归类为“司机”或“乘客”。不过,仅突变本身可能不足以导致癌症的发生,仍需要一个合适的微环境来促成完全癌变。以此类推,与炎症反应、免疫系统缺陷或生物失调相关的非致突变因素可能也是创造促衰老微环境所必需的。
时至今日,尽管没有明确的因果证据表明基因组不稳定性及随后对突变的修复与衰老过程直接相关,但其作为绝大多数癌症的特征这一观点已被学界广泛接受。多项研究表明,DNA修复缺陷可能导致衰老或癌症,DNA修复机制的损伤加速了小鼠的衰老,并与多种人类早衰症有关。有趣的是,许多早衰症(如共济失调毛细血管扩张症、范科尼贫血、布鲁姆综合征、科凯恩综合征、成人早衰症和着色性干皮症)患者也更易患癌。相对地,携带BRCA1或BRCA2突变的女性由于DNA修复缺陷更易患乳腺癌和卵巢癌,其乳腺上皮细胞表现出加速衰老的表型。此外,对人类和其他长寿物种的研究表明,DNA修复机制的增强与寿命的延长具有高相关性。
除了核DNA外,线粒体DNA(Mitochondrial DNA, mtDNA)也是外源或内源性压力源的靶点,这些压力源会导致mtDNA的突变和缺失,从而引起衰老和癌症。缺少DNA聚合酶γ的小鼠表现出加速衰老和寿命缩短的特征,这主要与mtDNA的缺失有关,而许多人类疾病也主要由mtDNA的损伤导致,这些都是mtDNA突变参与衰老及衰老相关疾病的直接因果证据。线粒体损伤还通过促进基因突变和肿瘤代谢物的产生导致癌症的发生,而肿瘤代谢物又通过代谢重编程和线粒体动力学的改变促进癌症的进程。
总之,这些发现支持了基因组不稳定性是衰老和癌症的元特征这一观点。同时,这些发现也揭示了,以减少DNA损伤或增强其修复或稳态维持机制为目标的干预措施可能会延缓衰老相关疾病及癌症的发展。
表观遗传改变及重编程
导致衰老和癌症的大量表观遗传改变包括DNA甲基化模式的改变、组蛋白翻译后修饰、染色质重塑和非编码RNA(Non-coding RNAs, ncRNAs)的功能变化。这些变化影响基因表达和其他关键的细胞过程,并促进衰老相关疾病和癌症的病理过程。
DNA甲基化
伴随着机体衰老,人体DNA表现出整体甲基化的降低,同时也表现出一些抑癌基因的过度甲基化。同样,人类癌症的表观遗传图谱表现出DNA甲基化模式的大规模重编程。与衰老的情况类似,癌症中绝大多数的DNA甲基化模式改变主要影响内含子或基因间区,但其中的一些甲基化改变会导致特定抑癌基因(如编码p16和p53的基因)的甲基化及沉默,从而对癌症的发生和发展起决定性作用。针对表观基因组改变的干预可能会延迟表观遗传时钟,同时也能抵抗血液系统恶性肿瘤的发生。一些甲基化事件有利于衰老和癌症的发生,为了对这些甲基化进行更高特异性和效率的干预,揭示在人类甲基化过程中整体和特异性变化发生的驱动因素是极其重要的。表观遗传调节因子DNMT3A(催化从头甲基化)和TET2(启动去甲基化)在众多干预靶点中脱颖而出,这主要是由于它们在意义不明的克隆性造血(Clonal hematopoiesis of indeterminate potential, CHIP)中频繁突变,是血液病和冠心病的危险因素。
组蛋白修饰
衰老细胞和癌症细胞在其组蛋白翻译后的表观修饰中表现出组织依赖性的变化,这可能导致转录改变、代谢失调和细胞稳态失衡。一些组蛋白去乙酰化酶(包括sirtuin家族的酶)与衰老和癌症相关,因其活性降低会导致染色质松弛,从而使DNA更易损伤。组蛋白去甲基化酶也与衰老和癌症相关。总而言之,这些发现为以组蛋白修饰酶为靶点治疗衰老相关疾病及癌症提供了理论支持。
染色质重塑
染色质重塑因子,如异染色质蛋白1α(Heterochromatin protein 1α, HP1α)、SWI/SNF家族和多梳蛋白等,可延缓衰老和癌症。它们的功能缺陷会扰乱染色质结构,这会导致衰老和癌症细胞中常出现的异染色质整体水平丢失和重新分布。例如,尽管SWI/SNF改变的促衰老和促癌作用的具体分子机制目前仍不清楚,但是高达25%的人类癌症中含有这种变化。
非编码RNA
ncRNAs,又包括lncRNAs、microRNAs(miRNAs)和环状RNA,主要通过转录后靶向长寿和致癌的分子通路的多组分来影响衰老和癌症。在细胞和动物模型中进行的功能获得和丧失研究证实了ncRNA,特别是miRNA与衰老和癌症的因果关联。例如,小鼠体内miR-455-3p的缺失会引起认知损害并缩短寿命,而其在人类癌症细胞中的下调则会促进癌细胞的增殖和侵袭活动。反之,该miRNA在小鼠体内的过度表达可以保护神经功能并延长寿命,其在异种移植实验中也被证实可以抑制肝癌的生长。另一个例子是,DICER1的磷酸化突变(是介导miRNA效应的RNA干扰机制的核心组成部分)在小鼠模型中加速衰老并促进癌症的发展。
炎症
衰老过程中会出现炎症的增加,此时机体进入一种被称为“炎症衰老”的状态,导致如骨关节炎、动脉粥样硬化、肌少症和神经炎症在内的许多衰老相关疾病。同时,炎症也是促癌特征之一。
炎症衰老源于多种分子、细胞和器官缺陷的协同作用,这些缺陷由衰老的其他所有特征引发。基因组的不稳定性会触发克隆造血和具有促炎表型的髓系细胞的增殖,从而导致心血管衰老。促炎性蛋白的过表达可能由表观遗传改变、蛋白稳态丧失和巨自噬失能引起,而这都是衰老的主要特征。过度的营养信号也有利于炎症的出现,这些信号激活GH/胰岛素样生长因子1(Insulin-like growth factor 1, IGF1)/PI3K/AKT/mTORC1轴,并导致作为衰老拮抗性特征的营养感应通路失调。此外,衰老细胞积累导致的慢性炎症通过衰老相关分泌表型(Senescence-associated secretory phenotype, SASP)典型的促炎细胞因子的过度产生而加剧炎症衰老。衰老相关的髓系和淋系祖细胞的耗竭阻碍了针对新抗原的有效免疫反应。T细胞类群的变化导致如下三点:(1)促炎性TH1和TH17细胞的增加;(2)免疫监视功能的缺失;(3)在整个生命周期中,自身耐受力降低以及随后出现的自身免疫性疾病的发病率增加。最后,由于维持生物屏障的能力减弱,尤其是肠道屏障的紊乱,会引起炎症加剧,这也将导致生态失调。
这些机制也在慢性炎症中起作用,而慢性炎症往往是癌症发生和发展的基础。肿瘤微环境中存在的炎性细胞通过促进(表观)遗传不稳定性、增殖、血管生成、代谢重编程、细胞外基质重塑、诱导侵袭和转移、癌症干细胞的维持以及阻断免疫监测,从而在癌症发生的各个阶段起到促进作用。
炎症和免疫系统的基因操作能够延缓不同组织和器官的衰老。敲除NLRP3或过表达SIRT2(其编码的蛋白具有去乙酰化和抑制NLRP3的功能)可改善衰老造血干细胞的功能和再生能力,并抑制衰老相关的炎症和胰岛素抵抗。此外,在小鼠模型中,通过阻断IL-1β、TNF-α、IFNAR1、caspase-1和NLRP3等方式进行抗炎治疗也可以延缓衰老。平行研究表明,肿瘤相关炎症细胞可以作为治疗靶点,例如,抑制它们在肿瘤内的募集、消耗、它们抗肿瘤作用的功能重启,或药理学阻断IL-1β和其他细胞因子。有临床研究发现,IL-1β阻断剂可以预防老年人非小细胞肺癌的发生。这种炎症导向的癌症治疗方法具有强大的潜力,可以与当前的化疗和免疫治疗策略兼施并行。
生态失调和多态的微生物组
肠道微生物群通过参与消化膳食营养素、抵御病原体、产生多种生物活性代谢物等多种生理过程,并向大脑和其他远端器官发出分子信号,从而对宿主健康的维持施加强有力的影响。这种肠道微生物-宿主双向通讯的中断会引起生态失调,并增加衰老相关疾病与癌症的风险。最新研究表明,生态失调和多态的微生物组对衰老和癌症的病理过程起着至关重要的作用。
衰老和癌症过程中微生物群的改变
肠道微生物群的多样性主要在青少年期建立,在成年后保持相对稳定。然而,在衰老过程中,微生物生态系统的组成和活性不断发生变化,最终导致其多样性大幅降低。随着衰老,微生物群在不同个体中出现特异性的变化,同时也呈现某些共同的特征,如参与炎症和免疫调节的微生物代谢产物的增加。有趣的是,在健康的百岁老人群体中,核心菌群(如拟杆菌属)减少,同时其他几个属(如阿克曼菌属)增加,而这些菌属被报道具有促进健康长寿的作用。百岁老人的肠道微生物群中还富含能够产生独特的二级胆汁酸的细菌,其对艰难梭菌和粪肠球菌等病原微生物具有强大的抵御作用。基于此,胆汁酸代谢可能有助于肠道内稳态的维持,并降低衰老相关慢性疾病的易感性。
平行研究表明,在癌症患者中肠道微生物群的组成显示出许多与疾病相关的变化。例如,基因毒性pks+大肠杆菌通过大肠杆菌素介导的突变诱导结直肠癌,而具核梭杆菌在结直肠癌组织中广泛存在,其可降低患者对化疗的临床反应。从机制上讲,肠道微生物群失调可能通过降低抗肿瘤免疫监测的有效性而诱发癌症。此外,肠道微生物群还可以影响免疫检查点抑制剂(Immune checkpoint inhibitors, ICIs)的疗效。值得一提的是,诸如嗜黏蛋白阿克曼菌等的与抗肿瘤免疫反应相关的细菌,也对机体的整体健康具有有利的影响。相对地,微生物群许多与癌症相关的变化也发生在其他非恶性疾病中,这表明肠道微生物群在不同疾病中的病理性变化是一致的。
肠道微生物群的延长寿命和抗肿瘤干预
异体粪便微生物群移植(Fecal microbiota transplantation, FMT)通过重置肠道微生物生态系统的组成,为延长健康寿命开辟了新道路。例如,在两个加速衰老的小鼠模型中,从野生型到早衰症小鼠的FMT提高了早衰症小鼠的健康寿命及实际寿命。值得一提的是,仅给小鼠施用粘蛋白A就足以获得类似的延长寿命效果。FMT还证实了肠道生态失调在全身性的慢性炎症以及衰老相关疾病引起的宿主免疫系统功能下降中发挥作用。科学家还开发了针对肠道细菌组成的其他干预措施,旨在使肠道微生物群重新年轻化,例如,向早衰症小鼠施用益生菌植物乳杆菌GKM3、在老年小鼠和猕猴中恢复足够水平的产生短链脂肪酸的细菌以及健康饮食(改善微生物群组成)。
特定的微生物对癌症的生长和进程具有抑制作用,这为新的癌症预防和治疗策略指明了道路。如前所述,嗜黏杆菌被报道对宿主新陈代谢的具有有益影响,且与癌症免疫治疗中对检查点抑制剂的阳性反应相关。粘蛋白A的免疫调节活性可能与其细胞膜中存在的支链二酰基磷脂酰乙醇胺相关,其优先诱导特定的细胞因子并重置免疫信号的激活阈值。此外,FMT已被证明可以改善黑色素瘤患者对免疫疗法的响应。总之,这些结果强调了生态失调、衰老与癌症间的紧密联系。
抑制癌症发生的衰老特征
衰老的两个特征具有显著的抑癌作用,该作用适用于端粒酶损耗,它能够阻碍癌细胞复制的永久性,而干细胞耗竭能够限制恶性肿瘤细胞的表型可塑性(图2)。然而需要注意的是,端粒酶损耗和干细胞耗竭都不能对抗癌症生物学的所有方面。
端粒缩短抑制细胞的无限复制
据报道,DNA损伤会影响染色体的末端(端粒)从而导致老化,因为复制性DNA聚合酶无法完成端粒区的复制。如果没有启动特定的对策,连续的细胞分裂周期会使得端粒渐进性缩短,以基因组不稳定而告终,最终导致永久的细胞周期阻滞(衰老)或细胞死亡。端粒损耗可以被端粒酶的逆转录酶活性所阻滞,或者被端粒的替代延长(Alternative lengthening of telomeres, ALT)阻滞。ALT是一种不依赖端粒酶的同源定向修复机制,在端粒酶缺陷的癌细胞中发挥作用。端粒酶仅在生殖细胞和干细胞中表达,而在分化细胞中通常不表达。在人类中,由基因决定的端粒酶缺陷与多种节段性早衰疾病有关,如再生障碍性贫血、先天性角化不良和肺纤维化。通过基因治疗激活端粒酶对再生障碍性贫血、心肌梗死和肺纤维化的小鼠模型具有治疗作用,并证实端粒和端粒酶在年龄相关疾病中的促进作用。人类数据的荟萃分析未能提供令人信服的证据以支持与生物学年龄相关的端粒长度减少。因此,虽然关于端粒在正常衰老中的作用仍然缺乏证据,但端粒酶(可能介导端粒独立性效应)在几种组织特异性的、模拟早衰的疾病中的参与似乎是可靠的。
限制大多数体细胞无限复制能力的倒计时机制的想法具有内在的启发式价值。在这种情况下,端粒损耗阻碍肿瘤的发生和发展似乎是合理的。显然,癌细胞必须重新激活端粒维持机制,无论是端粒酶(占所有癌症的80%-85%)还是ALT(在10%-15%的恶性肿瘤中,尽管更常见的是在间充质和神经上皮来源的肿瘤中),以实现复制的永久性,从而获得恶性肿瘤的主要特征之一。事实上,人类端粒酶逆转录酶(Human telomerase reverse transcriptase, hTERT)编码基因的启动子突变(-124 C>T和-146 C>T)是最常见的泛癌症驱动点突变之一,它通过招募ETS转录因子家族而使得hTERT不再沉默。在人成纤维细胞中过表达hTERT足以诱导其无转化的永生化,而端粒酶的重新激活似乎对人类细胞转化至关重要。正在开发的多种药物直接抑制hTERT(如BIBR1532)或属于端粒酶复合物的一种RNA模板分子hTERC(如inmetelstat和其他反义寡核苷酸)。这些药物通过核苷类似物或G-四联体稳定配体间接阻断端粒延长,或诱导针对hTERT的特异性免疫反应。然而,除了inmetelstat(一种端粒酶抑制剂)之外,这些方法都没有被证实具有临床活性,而inmetelstat对复发/难治性骨髓纤维化的疗效尚也未得到证实。UV1是一种基于DNA的疫苗,编码一种与泛素融合的hTERT失活形式,这可能在部分黑色素瘤患者中诱导具有治疗意义的免疫反应。关于ALT,抑制这一过程的分子都不是真正ALT特异性的,这意味着它们经常干预其他DNA修复过程。因此,到目前为止,还没有特异性的ALT抑制剂被引入临床。
端粒酶抑制剂在临床上有限的成功可能反映了端粒缩短和抑制癌症发生之间并不是完全相关的事实。因此,端粒酶缺陷的小鼠患有自发性恶性肿瘤的发病率增加,这在p53缺失的背景下尤为显著,尽管这种缺陷确实减少了肿瘤抑制基因Cdkn2a缺陷小鼠的癌症发生。在前瞻性人群研究中,循环白细胞中的短端粒与较高的患癌风险相关。此外,在急性早幼粒细胞白血病(Promyelocytic leukemia, PML)患者诊断时发现,短端粒与患者的不良预后表现出相关性。因此,端粒缩短和复制永久性之间的对立可能没有人们普遍认为的那么明显。这可能与端粒酶损耗本身导致基因组不稳定性有关,并表现为染色体融合和易位。
另一个难题来自于端粒酶不仅参与端粒的维持,还具有多种额外的功能。hTERT蛋白作用于线粒体,抑制细胞凋亡的内源性途径,同时也作用于mtDNA保护其基因组完整性,并与染色质重塑因子(如SMARCA4)和转录因子(如NF-κB)相互作用反式激活参与肿瘤发展的基因。hTERT也在功能上与FOXO1相互作用,反式激活催化NAD+生物合成的烟酰胺磷酸核糖转移酶和催化NAD+还原为NADH的甘油醛-3-磷酸脱氢酶,从而提高细胞生物能。基于这些见解,如果端粒酶的完全药理抑制能够实现,可能具有比以往认为的更大抗癌活性。对端粒酶的抑制是否会通过作用于患者骨髓、肺或皮肤中存在的干细胞从而加快其节段性衰老,这是另一个有待探索的谜题。
干细胞耗竭抵抗表型可塑性
衰老过程中的多个特征,包括端粒缩短、表观遗传改变、蛋白质稳态丧失、巨自噬失能、线粒体功能障碍和细胞衰老均减弱干细胞功能。然而,衰老的一个显著标志是干细胞耗竭,损害组织修复和更新,这在幼年状态下也可能发生。事实上,在年轻的组织中,损伤很容易激活和扩大预先存在的干细胞,甚至可以导致非干细胞的去分化,从而重新激活正常沉默的胚胎和干性转录程序,因此获得了参与组织修复的可塑性。组织修复需要通过分泌细胞因子、生长因子和细胞外基质的调节物来重塑微环境,这些物质共同作用有利于来自不同组织细胞的去分化和可塑性。这些损伤诱发的可塑性细胞可能获得多能祖细胞特征,从而用其实质和基质细胞类型取代丢失的细胞,重建功能性的超细胞单位,以重塑组织结构。然而,在老化的组织中,这种允许去分化和再分化的可塑性能力降低,这一缺陷可以通过转录因子OCT4、SOX2、KLF4和MYC(OSKM)的瞬时转基因表达将成体细胞转化为胚胎多能细胞(称为诱导多能干细胞或iPSCs)来修复,并提高受损老化组织的修复能力,如在大脑、胰腺、心脏、神经纤维、视网膜、肝脏、骨骼肌和皮肤中所表现的那样。值得注意的是,只有当OSKM转录因子以可控瞬时的方式表达时才会产生如此理想的结果,它们的过度激活可导致肿瘤发生。
癌症的特征之一是表现出表型的可塑性,即细胞在受影响的组织中转化为癌症干细胞,因为它们不能经历正常的终末分化,而是经历去分化,表现出分化阻滞或明显的转分化。去分化的例子包括结肠癌发生(由于失去分化诱导转录因子HOXA5和SMAD4)、黑色素瘤发生(由于失去转录因子MITF)和胰腺神经内分泌肿瘤(由于在β细胞中失去分化诱导的miRNA)。阻断分化发生在急性PML(由于PML-RARα基因融合),急性髓系白血病(Acute myeloid leukemia, AML)(由于AML1-ETO融合),以及许多其它癌症类型中。转分化影响胰腺导管腺癌(转录因子PTF1α和MIST1丢失导致腺泡细胞转分化,从而有导管细胞表型),雄激素阻断疗法抗性前列腺癌(SOX2有利于转分化为神经内分泌细胞状态),以及基底细胞癌(恶性细胞的转录组从与毛囊膨大干细胞的转录组相似转变为与滤泡间表皮中的基底干细胞转录组相似)。
据推测,与衰老相关的干细胞衰竭,由于其固有的可塑性降低,降低了细胞遗传积累和表观遗传改变的风险,以逃脱其正常的终末分化命运,从而获得恶性肿瘤的特征。然而,在此阶段,这个猜想在很大程度上是理论性的,需要进一步的实验证实。此外,在某些情况下,干细胞耗竭甚至可能促进癌症的发展。因此,造血干细胞池耗竭导致年龄依赖性的免疫缺陷,可能通过削弱免疫监测间接促进来自非造血细胞的肿瘤发生。
自噬失能:致癌和抑制肿瘤的作用
巨自噬(文中称之为“自噬”)是唯一允许清除多余大蛋白聚集物和受损胞质细胞器的细胞机制,从而确保它们的质量控制、清除和替代或“再生”。因此,基线自噬是维持细胞和机体健康所必需的;通过基因操作抑制其作用,如在小鼠体内全身诱导敲除或敲低重要的自噬基因Atg5或Atg7,显著加速细胞自主衰老和多种组织的炎症,从而缩短健康寿命并导致过早死亡。相反,通过转基因过表达Atg5或Becn1的功能获得性突变来刺激自噬,可以延长小鼠的健康寿命和实际寿命,同时降低肿瘤的发生率。使用这种小鼠模型进行实验,结果表明自噬对癌症生物学有双重影响,这可以在多西环素诱导的shRNA靶向Atg5的可逆自噬抑制模型中得到最好的说明。在这个模型中,通过不断补充多西环素永久抑制自噬,加速了生化、组织病理学和宏观上的衰老迹象,而不会诱导肿瘤的形成。然而,如果自噬抑制是暂时的(从出生后的3个月到7个月),自噬的恢复会在某些组织(肝脏和肾脏)上逆转机体虚弱和衰老的组织学迹象,而在其他组织(心脏或肌肉)则无法实现。然而,最重要的是,暂时的自噬抑制后,小鼠表现出通常在老年野生型小鼠中发现的癌症(如淋巴瘤和AML)以及通常在这类小鼠中不存在的恶性肿瘤(如肝细胞癌和骨肉瘤)的发病率增加。因此可以得出结论,暂时而非永久的抑制自噬是致癌的。这个有趣的悖论可以用一个假设来最好地解释,即自噬对癌症生物学具有双重影响:(1)自噬作为癌症启动的抑制因子和(2)自噬作为肿瘤发展的促进因子(图3A)。
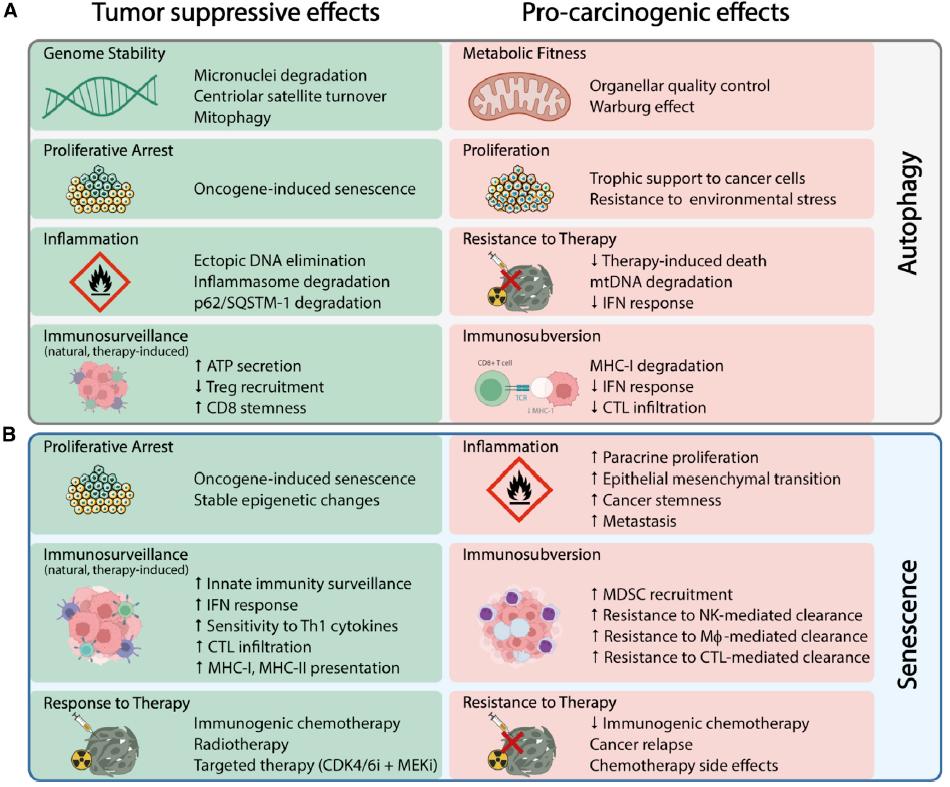
图3. 自噬和衰老在老化和癌症中的矛盾作用。
示意图为自噬(A)和衰老(B)的肿瘤抑制(左)和促进(右)功能(包括细胞自主和非细胞自主机制),归因于其在肿瘤发生、发展和对于抗癌治疗的反应/抵抗性作用。ATP,三磷酸腺苷;Tregs,FoxP3调节性T细胞;mtDNA,线粒体DNA;IFN,干扰素;CTLs,CD8+细胞毒性淋巴细胞;MHC,主要组织相容性复合物;MDSCs,骨髓源性抑制细胞;NKs,自然杀伤细胞;Mφ,巨噬细胞。
自噬的肿瘤抑制作用
在杂合子Becn1基因敲除或肝脏特异性嵌合基因Atg5敲除的小鼠中,部分抑制自噬促进自发性癌症发生。此外,通过组织特异性敲除Atg5或Atg7来完全抑制自噬,加速了由KrasG12D驱动的肺癌发生,特别是在p53缺失的背景下。相反,在这个模型中,自噬的药理刺激可以推迟肿瘤的发生。自噬抑制癌症发生的机制可能有多种,涉及细胞自主和炎症/免疫机制(图3A)。
在细胞自主水平上,基线自噬是细胞内稳态所必需的,例如,通过消除受损的线粒体(可以产生DNA损伤的ROS或释放其它促炎分子)并维持细胞三磷酸腺苷(Adenosine triphosphate, ATP)水平,以实现有效的DNA修复。自噬破坏的情况下,异常有丝分裂产生的微核,会扰乱细胞分裂,产生四倍体癌细胞前体。此外,自噬能够靶向中心粒组分,以维持中心体的稳定性和平衡分离,从而保障有丝分裂的准确性。因此,抑制自噬可能通过促进染色体和基因组的不稳定性来促进癌症发生。对于一些由衰老(比如由HRASV12和BRAFV600E引起的癌基因诱导衰老)和细胞死亡(比如在细胞铁死亡背景下)引起的细胞自主肿瘤抑制,自噬是必需的(图3A)。
在非细胞自主水平上,自噬有助于抑制促瘤性炎症,同时有利于癌症免疫监测(图3A)。为了阻止促炎通路的激活,自噬消除了异位胞质DNA(受损的线粒体或细胞核可释放这些DNA)。抑制炎症也可以通过选择性降解炎症小体组分和选择性自噬接头蛋白Sequestosome-1(SQSTM1/p62)来实现。在应激和死亡的癌细胞中,自噬有利于溶酶体分泌ATP,细胞外ATP在吸引树突状细胞和细胞毒性T淋巴细胞(Cytotoxic T lymphocytes, CTLs)进入肿瘤着床位中发挥决定性作用,同时减少局部免疫抑制调节性T(Regulatory T, Treg)细胞的存在。事实上,在人类乳腺癌中,自噬障碍的免疫组化检测与较低的CTL/Treg比值和较差的预后相关。而且,刺激自噬可以改善小鼠免疫原性化疗的疗效。这依赖于癌细胞的自主效应,但也可能涉及自噬驱动的功能性T细胞记忆干细胞池的维持,从而避免抗癌免疫反应的耗竭。
自噬的肿瘤促进作用
根据反复发生的情况,一旦出现潜在的致瘤克隆,自噬必须(重新)激活,以提高细胞的适应性,并促进向恶性肿瘤的过渡。因此,在多种情况下,基因或药理抑制自噬可以在小鼠模型中限制肿瘤发展。
自噬增加了细胞对不良条件的抵抗力,无论是内源性的(如缺氧、营养支持减少)还是医源性的(化疗或靶向治疗),这意味着它提供了对细胞死亡的抵抗作用(图3A)。自噬通过细胞自主方式增强细胞器质量控制来提高癌细胞代谢。此外,肿瘤中存在的基质细胞(如成纤维细胞)的自噬可能提高对恶性肿瘤细胞的趋向性支持。因此,自噬允许癌细胞增加其对恶劣的内源性条件(如缺氧、营养匮乏、营养因子缺失或被免疫效应物攻击)和抗癌化疗药物、放疗或靶向治疗的抵抗力。在特定情况下,恶性肿瘤细胞的自噬可能破坏免疫监测,例如,降解主要组织相容性复合物(Major histocompatibility complex, MHC)I类分子或抑制放疗诱导的癌细胞免疫识别所必需的1型干扰素(Interferon, IFN)反应。据报道,发生在肿瘤宿主(特别是肝脏)的自噬抑制,可促进肿瘤中1型和2型IFN反应,上调癌细胞中的MHC I类分子,避免T细胞耗竭和Treg的消除,来提高抗癌免疫反应(图3A)。
鉴于这些结果,有人建议使用自噬抑制剂治疗晚期癌症患者,特别是羟氯喹。尽管尚未得到证实,但已报道了羟氯喹联合Gemcitabine/Nab-pactitaxel治疗胰腺癌和联合Dabrafenib/trametinib治疗黑色素瘤的临床成功应用。然而,一项涉及自噬诱导剂ABTL0812治疗晚期实体肿瘤患者的临床试验也声称有了初步的抗肿瘤疗效,强调了自噬在肿瘤发展中的矛盾作用。在停止治疗后,使用比羟氯喹更有效和更特异的药物抑制自噬是否会导致与促衰老作用相关的机制或有利于恶性肿瘤发展的免疫抑制副作用仍有待观察。
细胞衰老及其在肿瘤形成中的矛盾性功能
术语“衰老”定义为响应有害(如DNA损伤)或无害(如发育信号)的各种非凋亡细胞状态。尽管衰老细胞具有高度异质性,但却展现出相同的表型属性,包括通常不可逆的无法跨越或重新进入细胞周期(伴有稳定的表观遗传重排)和旺盛的分泌表型(简称SASP)。正如在体内所见,衰老会引起相互矛盾的影响,会随着刺激强度,疾病分期和年龄而不断变化。衰老细胞在组织中短暂出现,然后是免疫细胞介导的清除,促进再生愈合,并且消除肿瘤前细胞变体。相反,衰老细胞的逐渐积累(由缺陷的免疫监视功能促进)加剧了组织纤维化和低度炎症现象,这定义了衰老过程并且提高了肿瘤转化的风险。在转基因小鼠模型中基因敲除p16+细胞足以延长自然衰老小鼠的寿命并降低包括肿瘤在内的年龄相关病变表征。在病理生理学层面证实了衰老的多效拮抗作用,在肿瘤生物学的几乎所有方面,促癌和抑癌作用都与衰老反应相关。这些矛盾效应背后的原因主要在于衰老细胞产生的可溶性介质的多样性——这反过来又受到起源的细胞类型影响——以及SASP对肿瘤生态位不同成分的可变影响(图3B)。
细胞衰老的抗肿瘤作用
致癌基因诱导衰老(Oncogene-induced senescence, OIS)是由于致癌基因(如Ras、BRAF V600E和c-Myc)的异常激活或肿瘤抑制基因(如PTEN)的丢失从而使得肿瘤暂时的处于癌前状态(图3B)。值得注意的是,OIS体系的成功建立需要较强的自噬能力。衰老阻止肿瘤发生的能力不仅仅是能够稳定诱导增殖停滞,并且还与免疫检测(前)肿瘤细胞的能力息息相关。根据这一概念,由CD4+ T细胞产生的TH1细胞因子——由携带突变NRAS G12V基因的衰老肝细胞招募——为巨噬细胞(Mφ)依赖的恶性前体细胞清除奠定了基础,并在绕过OIS的肿瘤中恢复衰老表型。此外,再激活p53依赖的衰老可以促进p53缺陷肿瘤的消退,SASP辅助天然免疫细胞(自然杀伤[Natural Killer, NK]细胞和Mφ)聚集在肿瘤处可以促进肿瘤的消退过程。虽然肿瘤发生需要破坏OIS,但肿瘤的衰老过程可以在常规化疗药物或靶向(如CDK4/6抑制剂)抗癌方案治疗后恢复,从而引起部分恶性细胞的衰老。因此,组合干预方案(例如,MEKi + CDK4/6i)旨在最大限度地提高肿瘤衰老水平,引发强力的NK介导的抗癌监测。此外,MEKi + CDK4/6i介导的衰老诱导了依赖于SASP的血管重塑,这足以增强CD8+ T细胞在胰腺导管腺癌微环境中的募集能力。在这种情况下,在MEKi + CDK4/6i方案中加入抗PD1免疫治疗,改善了对肿瘤生长的控制,从而验证了该方案的临床可行性。
衰老的促肿瘤作用
慢性、无菌性炎症能导致肿瘤发生,这一概念解释了衰老过程中的一些致瘤特性(图3B)。消除中年小鼠p16+衰老细胞可以降低肿瘤形成率。在培养过程中,衰老成纤维细胞产生的炎症细胞因子刺激癌前和恶性上皮细胞的增殖,并诱导上皮-间充质信号转变。值得注意的是,衰老成纤维细胞与癌细胞共注射加速了肿瘤异种移植瘤的发生和发展。
衰老在肿瘤发生过程中有很大作用,衰老的垂体胚胎前体表达致癌性β-连环蛋白,这提高了体内逃避OIS屏障的肿瘤前体细胞的增殖速率。重要的是,衰老的癌细胞或非转化的衰老基质细胞可以释放SASP因子,这些因子使得肿瘤微环境中免疫功能受阻,为肿瘤发展奠定了基础。相一致的,许多证据表明,在肝癌、前列腺癌和皮肤癌的小鼠模型中,通过衰老细胞分泌因子(例如,IL-6、IL-8和CCL2),募集髓样衍生抑制细胞(Myeloid-derived suppressor Cells, MDSCs)从而导致肿瘤微环境中免疫抑制的发生。与这一观察结果一致的是,通过药物阻断MDSC募集可以抑制肿瘤生长,并增强衰老诱导化疗的作用。那些逃脱了免疫监测的衰老细胞也会增加转移的风险。例如,Neu和MMTV-PyMT乳腺癌模型中哺乳动物上皮诱导的衰老赋予上皮细胞干细胞特性,使其侵袭性和转移潜能增强。在甲状腺乳头状癌的小鼠模型中,位于集体浸润部位和淋巴结前部的衰老癌细胞产生了利于癌症侵袭的CXCL12梯度。此外,衰老的成骨细胞以IL-6依赖的方式驱动转移性乳腺癌细胞的骨生态位定植。衰老细胞也被假定介导对治疗的抵抗,同时导致与标准抗癌方案治疗相关的脱靶效应和癌症的复发。例如,残留的衰老癌细胞释放的嗜酸细胞活化趋化因子、CXCL5和CCL5可以抵抗阿霉素在MMTV-PyMT乳腺癌模型中的作用。在相同的模型中,全身清除阿霉素治疗产生的p16+细胞可以减少全身衰弱的迹象,并限制肿瘤的复发。值得注意的是,观察发现衰老的B细胞淋巴瘤细胞在人工诱导下,可以重新进入细胞周期并获得祖细胞特征,赋予它们发展为侵袭性肿瘤的能力,这是一种癌症中由衰老导致的终末停滞状态的例外。尽管如此,还需要更多证据来进一步在生理环境中验证这种效应。
基于此处列举的临床前实验证据,在大部分肿瘤中诱导衰老的方法具有进入临床实践领域的潜力,假设与辅助免疫疗法或衰老疗法结合,实现去除潜在有害的衰老逃逸变异体的目的,将具备更大的潜力。有趣的是,这种干预措施可能会产生更深远的影响,通过清除无癌器官中的衰老细胞,同时改善癌症患者的衰老相关表型。
衰老和癌症的共同代谢特征
营养感知的失调是一个公认的衰老特征,和能量代谢的重编程一起被纳入了检测癌症的指标。这两个特征表现出明显的共性,但衰老相关的营养感知失调和癌症相关的细胞能量代谢失调之间也有一些差异,甚至是拮抗的。这就需要对衰老和癌症过程中常见的代谢特征进行分析(图4)。
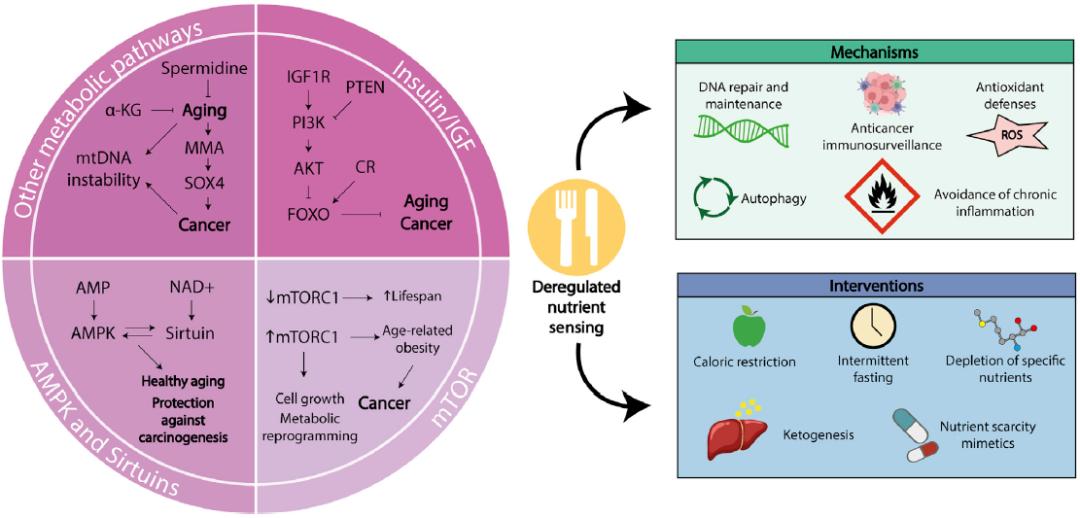
图4. 衰老和癌症的共同代谢特征。
展示了在衰老和癌症过程中的多个相互关联的营养感知系统中,胰岛素和IGF-1信号(IIS)轴,哺乳动物雷帕霉素靶蛋白(Mammalian target of rapamycin, mTOR)和AMP活化蛋白激酶(AMP-activated protein kinase, AMPK)通路,以及去乙酰化酶的Sirtuin家族。在衰老和癌症中,其他的代谢途径向相同的方向转变,比如亚精胺,a-酮戊二酸和甲基丙二酸(Methylmalonic acid, MMA)。这个图还包括基于靶向不同的代谢途径实现延缓衰老和抗肿瘤的饮食和药物干预。
营养感知网络是复杂的,因为它包括细胞外配体(即IGF和IGF结合蛋白[IGFs and the IGF binding proteins, IGFBPs],拮抗后者),与其互作的受体酪氨酸激酶,细胞内信号级联(即原癌PI3K-AKT和RAS-MEK-ERK通路)以及反式激活参与必要的细胞过程基因的转录因子(即FOXOs和E26因子)。在多个相互关联的营养感知系统中,胰岛素和IGF-1信号通路(Insulin and IGF-1 signaling, IIS)轴、mTOR和AMP活化蛋白激酶(AMPK)通路,以及去乙酰化酶的Sirtuin家族,在衰老和癌症的代谢失调中发挥重要作用。IIS途径体现细胞葡萄糖水平,是进化过程中最保守的衰老控制途径。在人类和模式生物中,降低该通路中心成员活性的遗传多态性、基因突变和抑制剂以及其下游细胞内效应物都与长寿有关。年龄相关代谢综合征包括全身代谢活性丧失和细胞类型特异性改变,包括例如主要影响肝脏和骨骼肌肉的IIS抵抗、增强的全身胰岛素水平(由胰腺中的β细胞产生)和循环性IGF-1的改变(主要由肝细胞产生)。IIS通路的下游效应因子是FOXO转录因子家族,它们也参与癌症,并介导卡路里限制(Caloric restriction, CR)对健康衰老的许多积极影响。例如,小鼠FOXO1参与了CR的肿瘤抑制作用,而其他IIS活性降低的模型,如癌基因PI3K的亚形态和PI3K拮抗癌抑制基因PTEN过表达的小鼠,它们出现寿命延长的现象。通过使用PI3K的显性阴性p110α亚型来抑制心脏IGF1R可以延长雄性小鼠的寿命,并改善老年小鼠的心脏功能。此外,对IGF1R的药理学抑制可以提高抗癌免疫监测,长期施用抗IGF1R抗体可以增加雌性小鼠的寿命并减少炎症和癌症发展。这些发现证明IIS轴是延缓衰老和抗肿瘤干预的一个重要靶点。
mTOR激酶是多蛋白复合物mTORC1和mTORC2的一部分,它们参与对高氨基酸浓度的感知,并调节合成代谢的多个方面。mTORC1活性水平较低的转基因小鼠寿命延长,并且S6K1(mTORC1主要底物)缺陷的小鼠寿命也有延长。此外,mTOR活性随着小鼠下丘脑神经元衰老而增加,并导致与年龄相关的肥胖,这是癌症的一个重要危险因素。mTOR在癌症中经常被激活,刺激细胞生长,促进适应性进化,并通过重新连接葡萄糖、氨基酸、核苷酸、脂肪酸和脂质代谢来促进肿瘤的代谢重编程。一些mTOR靶向药物,包括不同的雷帕霉素及其衍生物,已经用于扰乱癌细胞代谢,也作为老年保护策略的一部分进行探索。
AMPK和Sirtuins的作用方向与IIS和mTOR的作用方向相反,因为它们感知的是营养缺乏而不是营养丰富,而且刺激的是分解代谢而不是合成代谢。AMPK和Sirtuins分别通过检测高水平AMP和NAD+水平来感知低能状态。值得注意的是,AMPK和SIRT1参与了一个正反馈通路,它们将两个低能量状态的传感器变成单一的响应。总的来说,AMPK和几种Sirtuins的激活或上调有利于健康衰老,预防癌症发生。因此,活化的AMPK磷酸化可以消除参与细胞生长和维持癌细胞干性通路的关键成分,可以构成抗癌药物的潜在靶点。SIRT6是一种促长寿因子,其可通过抑制HIF1和MYC的转录从而抑制肿瘤。同样,SIRT6在人类癌症中经常发生突变和失活。类似的,SIRT7直接与MYC相互作用,减轻由该致癌基因激活引起的显著代谢改变。SIRT2去乙酰化BARD1以增强其与肿瘤抑制因子BRCA1的异源二聚化,从而通过同源重组改善BRCA1依赖的DNA双链断裂修复。
在衰老和癌症过程中,还有其他的代谢途径向同一个方向变化。因此,衰老可能与循环性亚精胺和α-酮戊二酸的下降有关,这两种物质都具有延缓衰老和抗癌的潜力。甲基丙二酸(MMA)是丙酸代谢的副产品,随着人类年龄的增长而在血浆中增加,诱导SOX4的表达,从而导致转录重编程,进一步增加癌症的侵袭性。在乳腺癌和肺癌细胞中,细胞内MMA随着甲基丙二酰辅酶A异构酶的下调而增加,从而减少丙酸盐介导的回补通量,这可能增强肿瘤细胞的转移潜能。基因组不稳定性影响mtDNA,此外其他机制也会损害衰老过程中的线粒体功能,而mtDNA序列的变化在肿瘤中很常见,这将使肿瘤的产能方式向糖酵解和谷氨酰胺解方向转变。
总的来说,这些发现与合成代谢信号促进衰老和癌症的假设是一致的,而营养感应信号的降低延长了寿命并抑制了癌症的发展,这可能是由于增殖和代谢速率的降低,从而减少细胞损伤导致的。因此,营养感知途径的不同组成部分都是促进长寿和抗肿瘤干预的潜在靶点。现有的饮食方案有很多,如旨在模拟营养素缺乏状态的药物治疗,以及基于连续CR、间歇禁食、生酮饮食(通过消除碳水化合物)或特定营养素耗竭的不同饮食方案(如蛋氨酸),科学家正在这方面进行积极探索,并已取得了令人欣喜的结果。在机制上,所有这些干预措施都集中在细胞应激反应的刺激源上,如DNA修复和维持、抗氧化防御、自噬、避免慢性炎症和抗癌免疫监测。然而,对癌症和衰老过程中的代谢变化机制的进一步了解,对于设计更有效的营养干预措施来说很有必要,这些干预措施具有综合的保护和抗癌作用。也就是说,个体癌症亚型的代谢特征是高度多样化的,由特定的致癌基因和通路驱动,这意味着许多特征可能与衰老过程中观察到的特征不一致。
免疫逃逸:衰老所致的肿瘤特征
肿瘤必须逃避免疫监视才能达到临床上的可检测水平,这意味着它们要么被动的选择“藏匿”免疫识别(免疫选择),要么积极的抑制免疫应答(免疫颠覆)。过去的十年间,肿瘤标志物变得越来越重要,反映着免疫治疗(尤其是针对PD-1和PD-L1之间相互作用的ICIs)已经逐渐成为了应对各种不同恶性肿瘤抗癌药物开发的骨干之一。恶性细胞的免疫逃逸可能反映了肿瘤-免疫应答中产生的特定进化特征,依据3E的假说,(前)恶性细胞最初被消除,然后达到一种平衡态,最后“突破”免疫逃逸,因为它们通过遗传和表观遗传适应逐渐改变了自身表型。因此,免疫逃逸与衰老标志物之间并没有明确的关系,可能只是与衰老细胞免疫清除失败和胞内通信改变相关。相反,许多的衰老标志物却有利于肿瘤的免疫逃逸,比如自噬失能,衰老,慢性炎症(图1),以及包括T淋巴细胞在内的免疫细胞衰老。
衰老组织中亚精胺进行性耗竭所致的自噬失能可能会因树突细胞无法识别自噬缺陷的肿瘤细胞、T淋巴细胞和NK细胞应答缺陷而干扰肿瘤细胞的免疫识别过程。因此,口服亚精胺可以在小鼠模型上重建肿瘤的免疫控制,在培养基中添加亚精胺可以在体外层面恢复老年供体来源B细胞和T细胞的应答缺陷。
细胞衰老可能是有利于肿瘤免疫逃逸的另一个因素。实际上,衰老细胞通常会被巨噬细胞和NK细胞清除,意味着存在免疫逃逸的选择性压力。在剥夺衰老细胞休眠,促进恶性转化的致癌过程中,产生的肿瘤细胞早已被预先选择以逃避免疫识别。此外,肿瘤微环境中衰老细胞的积累可能使系统偏于炎症和纤维化,进而导致局部免疫抑制和物理层面上阻碍T淋巴细胞接近恶性细胞,从而将肿瘤转变为免疫荒漠。实际上,具有选择性杀伤衰老细胞特性的药物,比如心脏糖苷和BCL2抑制剂Venetoclax可在免疫原性化疗和免疫检查点封锁的背景下提高肿瘤的免疫监视功能。
衰老相关的慢性炎症可通过加快细胞周转、削弱免疫监视从而显著促进肿瘤的形成。部分原因是由于肿瘤微环境中年龄驱动的免疫抑制细胞类型不断积累,比如MDSCs,2型巨噬细胞(Type 2 macrophages, M2),Tregs,以及由于细胞外基质的生物物理变化影响免疫效应物的迁移和位置。然而,除了这些常规的年龄相关特征,T淋巴细胞等免疫效应细胞也会单独或整体性的衰老。这涉及一系列机制,包括胸腺生成的T淋巴细胞数量减少、细胞内在改变(比如基因和表观遗传变化、线粒体功能障碍,自噬失能),并进一步导致了次要的功能变化(T细胞受体库减少、初始细胞和记忆细胞失衡、效应体可塑性下降和衰老)。总之,在缺乏治疗干预的情况下,这些变化可能会削弱自发的肿瘤免疫监视。然而,在临床前实验中,它们对靶向CTLA-4,PD-1,PD-L1的ICIs的抗癌功效却几乎没有影响。
老年癌症患者的治疗反应
通常认为老年癌症患者(通常指>60岁或65岁的个体)的临床治疗效果不如年轻对照组,常导致老龄个体在新的临床治疗方法中样本选取较少。然而这种假设过于简单化,因为它未能认识到癌症类型和特定患者群体之间存在的多重异质性,并且也未认识到不同年龄组之间治疗方法的差异,以及一些流行病学研究的方法问题,包括“老龄”的定义。
在急性AML中,60岁以下患者,以柔红霉素和阿糖胞苷为基础的化疗痊愈率在70%,但是60岁以上患者的痊愈率却只有45%-50%。而在80岁以上的弥漫性大B细胞淋巴瘤(Diffuse large B cell lymphoma, DLBCL)患者中,多模式化疗加上利妥昔单抗的治愈率则是从64%下降到了43%,这一定程度上(但不是全部)是由于患者虚弱所导致的治疗强度变化。相对于50岁以上患者,多发性骨髓瘤的治疗方法对50岁以下患者更为有效并增加了其总体生存率。同样,年龄小于64岁接受胃癌手术的患者也显著改善了疾病预后。然而,在接受手术外加辅助化疗的胃癌患者中,不同年龄组的结果之间没有差异,这可能反映着在年轻和健康个体中存在有相对非活性化疗滥用的情况。此外,与老年患者(>65岁)相比,上消化道癌的年轻患者(<45岁)总体生存率获得延长,而对于远端疾病和至少两个转移灶的患者则恰恰相反。
对于绝经后的ER+HER2-乳腺癌患者,年龄大于75岁的患者相较于年龄在55-75岁之间的患者,管腔B亚型患病率增加,预后更差。然而,年轻女性(<40岁)患三阴性乳腺癌(Triple-negative breast cancer, TNBC)的可能性最高(>75岁的女性最低),其侵袭性也比ER+HER-的对照组更加强烈,也和较差的疾病转归相关。年轻(<35岁)也是可手术乳腺癌患者中复发的一个风险因素,这表明年轻人和老年个体完全不同的发病机理。类似的考虑可以扩展到其他几种肿瘤情况。实际上,一个大型回顾性研究表明ICIs提高了黑色素瘤患者的总体生存率,但是对于60岁以上患者的提升幅度更为明显,尽管60岁以上患者接受ICIs的可能性更小。同样,小于45岁且被诊断为非小肺细胞癌(Non-small lung cell carcinoma, NSCLC)患者预后显著差于相对高龄的对照组。这种差异似乎反映了多种因素,包括吸烟情况、疾病诊断分期和就医延误。
虽然在不同年龄组之间,接受靶向抗癌药物或免疫治疗的转移性肾透明细胞癌患者的总体生存率似乎没有变化,但根据报道,50岁以下患者会缩短无进展生存期(Progression-free survival, PFS),而70岁以上患者的治疗毒性风险则会增加。相似地,年龄似乎并不会影响通过手术切除了肝癌的患者的总体生存率或无进展生存期,但是老龄患者发生2级甚至更高级别术后并发症的风险增加。对在胰腺癌患者队列中(以50岁为分界)的发现也表明,年龄因素对总体生存率的影响较小,尽管50岁以下的患者始终比老龄对照组接受更多的放疗或化疗。因此,至少在某些肿瘤条件下,主要影响标准护理疗法和疾病转归敏感性的并不是年龄。
肿瘤或宿主的免疫生物学参数对这些关联的实际贡献可能随年龄变化,然而,由于一些潜在的混杂因素,大多情况仍待进一步阐明(图5)。首先,在许多情况下,年龄会给治疗方式、持续时间和治疗强度造成相当大的改变,反应了老年个体普遍下降的身体机能。这在肿瘤适应症中最为突出,通常采用高强度的治疗程序,包括造血干细胞移植(Hematopoietic stem cell transplantation, HSCT)和剧烈的化疗。其次,一些最终可能影响治疗结果的行为可能会随着年龄的增长而改变。例如,老年人通常比年轻人更频繁地接受医疗照顾,特别是对于可能由肿瘤引起的相对轻微的症状(例如,肺癌患者持续咳嗽)。这可能产生这样一种情况,即年轻人从出现症状到诊断(因此开始治疗)的周数/月数可能比老年人高得多。第三,老年人的定义随着预期寿命的增加而演变,同一恶性肿瘤的不同研究可能采用不同的临界值来区分老年和年轻患者。最后,许多试图将癌症患者的年龄与治疗反应和疾病结局联系起来的研究都存在方法学问题,包括缺乏适当的统计评估(多变量回归分析)以及在缺乏疾病特异性生存(Disease-specific survival, DSS)信息的情况下,使用全因死亡作为总体生存率的指标。仔细考虑这些(以及潜在的其他)因素将使我们能够对老年人肿瘤免疫生物学及其对治疗的反应之间的联系获得更深入的了解。
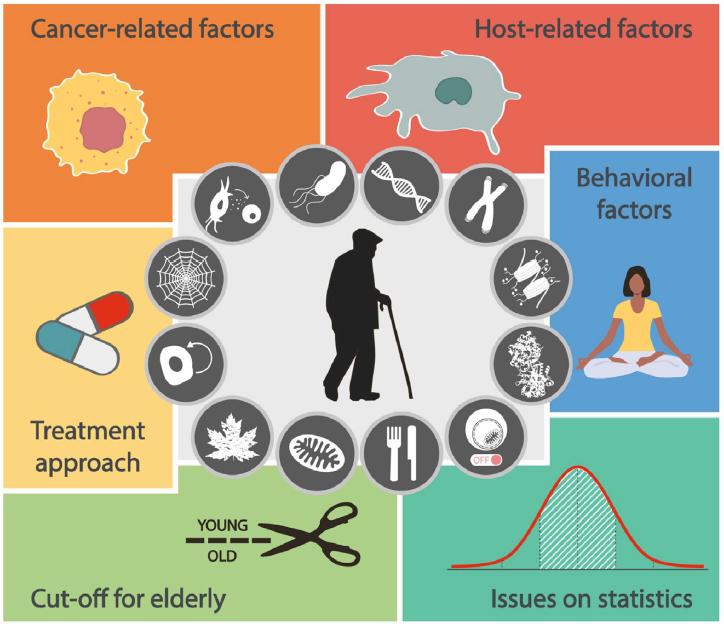
图5. 老年人癌症的治疗干预。
图中展示了一些影响治疗效果与衰老之间关系的多种因素。特别强调了可能的混杂因素,这些因素使得年轻和年老患者治疗效果的比较存在问题。
结论与观点
衰老和癌症之间错综复杂的联系,主要体现在日益失调的生物体内(前)恶性细胞的矛盾激增中,它们经历达尔文选择的过程,适应性逐渐提高。恶性细胞通常是在衰老相关的菌群失调、免疫监视功能衰退、炎症和代谢偏差等共同促进肿瘤发生的背景下,由于在基因组和表观基因组水平上细胞身份维持的失败而产生的。同时,癌细胞必须克服一些衰老相关的机制,这些机制通常限制细胞的适应性、增殖和可塑性,如自噬抑制、细胞衰老、干细胞耗竭和端粒缩短。
正如本文所述,衰老与癌症之间的关系非常复杂。一些衰老特征无疑会促进癌症(衰老与癌症的元特征),比如基因组不稳定性、表观遗传改变、慢性炎症和生态失调,这意味着对这些特征的预防性抑制(例如通过避免DNA损伤或刺激DNA修复、调节表观遗传酶、抗炎药物或从年轻供体到老年受体异时粪菌移植)可产生预防癌症的效果。
其他衰老特征主要起到抑制肿瘤的作用。这些拮抗特征包括端粒缩短以及干细胞耗竭,因为癌细胞必须(重新)激活端粒修复机制才能无限增殖,而恶性细胞必须恢复干细胞特征才可获得表型可塑性。因此,抑制端粒修复或促进干细胞耗竭可以被用于预防或治疗癌症,尽管代价是会加速衰老。然而这种推测目前还处于理论阶段,并没有在癌症治疗方面取得任何实质性进展。
其他衰老特征与癌症之间的关系更加模糊,如自噬抑制和细胞衰老。自噬是抑制肿瘤的,因为对它的暂时性抑制有利于细胞恶性转化和逃避免疫监视,但恶性细胞前体必须重新激活自噬来增加它们的适应性。这引发了两种相反的主张,即通过增强或抑制自噬来治疗癌症。细胞衰老以细胞自发的方式阻止初始癌变,但组织内衰老细胞的积累刺激了慢性炎症,将肿瘤转化为“不愈合的伤口”。因此,诱导细胞衰老和消除衰老细胞(Senolysis)都被提出作为可能的抗癌策略。
与衰老相关的营养感应失调可能为癌细胞的代谢重编程奠定基础,然而,这通常会激活一组由致癌基因和(表观)遗传扰动驱动的独立代谢变化,损害肿瘤抑制功能。因此,尝试逆转营养感应失调可能具有肿瘤预防作用,但对已确诊癌症的单独治疗方法可能效果不佳。
总的来说,衰老特征与癌症特征之间的这些相同、拮抗或矛盾的关系,可能解释了癌症与衰老之间特殊的流行病学关联,即恶性肿瘤的发生率在老年人中随着年龄的增长而增加,但在九十多岁和年龄更大的百岁老人中却下降。然而目前的生物学和临床知识并不足以理解年龄对治疗结果的影响,这意味着时序年龄(而不是生物学年龄)几乎不能被用作指导个性化医学治疗策略的一个标准。事实上,肿瘤学家很少或从不在他们的患者中研究生物学衰老时钟。然而在临床研究中测量这些时钟(例如甲基化时钟、CHIP、基因组异质性和正常组织活检中衰老细胞占比、循环代谢物和蛋白质、炎症生物标志物等)将有助于理解生物学年龄(而不是时序年龄)对治疗结果和患者预后的真正影响。最重要的是,肿瘤学家和老年医学专家之间的合作应该加强,不仅因为大多数癌症是在老年人中诊断出来的,而且因为肿瘤本身以及它的治疗都可以加速衰老过程。我们在本综述中提出的所有12个衰老特征和14个癌症特征都与从分子到个体(包括微生物群)的8个机体组织层次密切相关,构成了一个复杂而多维的相互作用网络,这可能有助于进一步理解和靶向衰老与癌症的新方法(图6)。未来的临床前研究和临床试验必须确定哪些延缓衰老的措施可以与抗肿瘤治疗安全地结合使用,而不会降低后者的效果。本文可能为这种延缓衰老-抗肿瘤的联合策略奠定了(部分)理论基础。
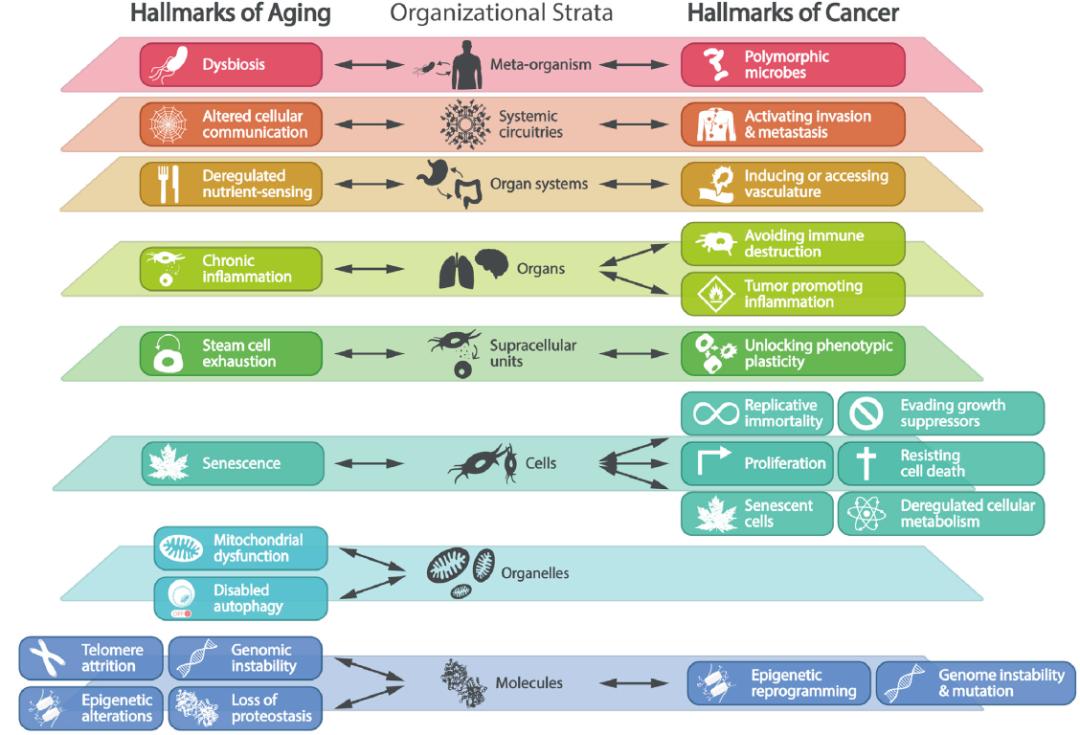
图6. 衰老和癌症特征在机体组织层次的整合。
本文讨论的12个衰老特征和14个癌症特征通过提出的八个机体组织层次相互联系,构成了一个复杂的相互作用网络,这可能为衰老和癌症的实验探索和治疗提供新的方法。

原标题:《【重磅综述】Cell重磅综述|全面解析衰老与癌症的关系》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

