- +1
【科技前沿】Protein & Cell 综述 | 甘波谊团队总结代谢性细胞死亡的机制以及癌症中的…
细胞死亡在维持生物体细胞的稳态中起着重要作用,然而这种微妙的平衡可能会在包括癌症在内的多种疾病中被破坏,因此细胞死亡抵抗是癌症的一个标志。细胞死亡大致可分为两种类型:意外细胞死亡和调节性细胞死亡。意外细胞死亡是由于物理或化学损伤等外部因素被动发生的,缺乏内部信号传导和执行机制。相比之下,调节性细胞死亡是由一系列细胞内信号传导和执行机制精心策划的高度控制和有序的过程。最近的研究表明代谢性细胞死亡是由细胞代谢不平衡导致的调节性细胞死亡新形式。回顾过去的十几年,科学家们对代谢性细胞死亡的机制研究取得了重要进展,为将其应用于癌症创新疗法的开发指明了方向。
近日,安德森癌症中心甘波谊教授团队在Protein & Cell上发表了综述文章Metabolic cell death in cancer: Ferroptosis, cuproptosis, disulfidptosis, and beyond 。本综述总结了代谢性细胞死亡(包括铁死亡、铜死亡、双硫死亡、溶酶体锌死亡和碱死亡)的机制,并探讨了它们在癌症治疗中的潜力。更重要的是,文章强调了代谢细胞死亡途径的复杂性,并为其应用于癌症的创新疗法提供了独特的见解。
 最近的研究发现代谢性细胞死亡是由某些营养素(例如葡萄糖和氨基酸)或金属(例如铁和铜)的超载或耗尽导致的细胞代谢失衡所引起。虽然代谢性细胞死亡在生物发育过程中的作用仍然存在争议,但对其复杂机制的深入了解在揭示疾病进展并为开发治疗各种疾病(特别是癌症)的新疗法方面具有巨大潜力。
最近的研究发现代谢性细胞死亡是由某些营养素(例如葡萄糖和氨基酸)或金属(例如铁和铜)的超载或耗尽导致的细胞代谢失衡所引起。虽然代谢性细胞死亡在生物发育过程中的作用仍然存在争议,但对其复杂机制的深入了解在揭示疾病进展并为开发治疗各种疾病(特别是癌症)的新疗法方面具有巨大潜力。铁死亡
铁死亡是一种由脂质过氧化物异常积累所引发的铁依赖性细胞死亡,它在形态学和机制上都不同于其他类型的调节性细胞死亡。例如,发生铁死亡的细胞不表现出染色质浓缩或 caspase-3裂解,而是表现出线粒体收缩和线粒体嵴减少。铁死亡的另一个定义特征是它被铁螯合剂或亲脂性抗氧化剂阻断,但不能被其他常见细胞死亡途径的抑制剂阻断。从代谢的角度来看,铁死亡表示一种细胞状态,其中促进脂质过氧化的代谢活动大大超过了旨在抵消这一过程的细胞防御系统的缓冲能力。哺乳动物细胞的细胞膜富含多不饱和脂肪酸(PUFA),而PUFA中的双烯丙基非常容易发生过氧化从而导致铁死亡发生。ACSL4促进游离PUFA连接到辅酶A形成PUFA-CoA,随后通过LPCAT3重新酯化并生成PUFA-PL。PL-PUFA的过氧化主要由非酶芬顿反应引发,该反应导致磷脂自由基的形成,引发链式反应并传播脂质过氧化。铁死亡的铁依赖性质凸显了铁在促进芬顿反应中不可或缺的作用,芬顿反应是脂质过氧化的驱动力,同时铁还作为参与脂质过氧化的酶(包括 ALOX 和 POR)的关键辅助因子。细胞铁稳态通过铁的吸收、储存、使用和输出过程的复杂协调受到严格控制,这种精细调节的任何干扰都会影响细胞内不稳定铁池的水平,从而影响癌细胞对铁死亡的敏感性。如转铁蛋白、乳转铁蛋白和转铁蛋白受体促进的铁摄取会刺激铁死亡,而转铁蛋白受体本身被确定为铁死亡标志物。铁相关的复杂机制共同影响细胞对铁死亡的敏感性,强调了铁在铁死亡发生中作为催化剂和调节器的多方面作用。
GPX4依赖性系统被广泛认为是细胞中防御铁死亡发生的关键。通过谷氨酸/胱氨酸逆向转运蛋白转运至细胞的胱氨酸被还原为半胱氨酸,随后生成谷胱甘肽(GSH)。GPX4利用GSH催化有害脂质氢过氧化物还原为无害脂质醇,从而有效抑制铁死亡发生。细胞中也存在着多种不依赖于GPX4的铁死亡防御系统,涉及多种自由基捕获抗氧化剂,例如泛醇、四氢生物蝶呤和还原形式的维生素K。质膜上的FSP1作为NADH/NADPH依赖性氧化还原酶发挥作用,利用NADH和NADPH的还原能力将泛醌转化为其保护性还原形式泛醇,从而独立于GPX4有效抑制铁死亡发生,同时还有研究表明FSP1在介导维生素K还原方面发挥着关键作用。泛醌合成及其随后还原为泛醇主要发生在线粒体内,是由位于线粒体内膜上的一系列酶(包括 DHODH 和 GPD2)所催化。相应地,DHODH或 GPD2的失活或缺失会使线粒体内泛醇的产生减少从而导致癌细胞铁死亡敏感性升高。一项研究发现,尽管线粒体 DHODH 和胞质酶(包括氨基甲酰磷酸合成酶 II、天冬氨酸转氨基甲酰酶、CAD和UMPS)定位在不同的亚细胞区室中,但它们可以通过线粒体外膜上的VDAC3形成被称为嘧啶体的复合物,促进底物通道并促进有效的嘧啶生物合成和铁死亡防御。四氢生物蝶呤被认为是芳香氨基酸羟化酶和各种酶的辅助因子,也是一种具有显着的铁死亡抑制能力的氧化剂。GCH1是一种催化四氢生物蝶呤生物合成途径限速步骤的代谢酶,其具有抑制铁死亡的作用(图1)。
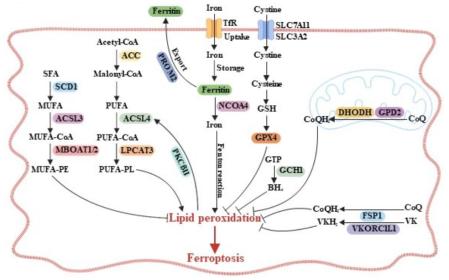 图1 铁死亡的关键机制
图1 铁死亡的关键机制在当前靶向铁死亡的肿瘤治疗临床前研究中,重点主要围绕几种I类铁死亡诱导剂(FIN)开展,即IKE、sulfasalazine、sorafenib和cyst(e)inase,但这些FINs在用作独立治疗时通常仅产生中等的抗肿瘤作用。有趣的是,传统疗法(例如放射和免疫疗法)与FINs通常会产生协同效应,通过促进肿瘤内铁死亡的诱导来提高整体治疗效果。此外,未来探索的另一个方向在于了解FINs治疗在肿瘤免疫微环境中的影响。FINs抑制免疫细胞活力的作用引起了人们的担忧,其可能会损害铁死亡诱导治疗的整体治疗效果。简而言之,虽然FINs可以有效地靶向肿瘤细胞,但必须考虑它们对免疫细胞的存活和功能的潜在影响,这种多方面的考虑强调了平衡肿瘤细胞破坏与免疫细胞保存的复杂性。
铜死亡
铜是一种不可或缺的微量营养素,作为催化辅助因子,在抗氧化防御、线粒体呼吸和重要生物分子的合成等一系列生物过程中发挥着重要作用。然而,过量的细胞铜水平可以引发一种独特形式的受调节细胞死亡,称为“铜死亡”。细胞内铜的水平通过铜的吸收、利用、储存和输出等过程的复杂相互作用受到严格的调节。细胞对铜的吸收主要由CTR1介导,而当CTR1缺乏时,DMT1也有助于细胞对于铜的吸收。随后COX17将细胞内的铜转运至 CCO,后者是线粒体电子传递链中不可或缺的组成部分,并依赖铜来执行其氧化磷酸化的基本功能。此外细胞内过量的铜也可以通过ATP7A和ATP7B输出。参与铜代谢的蛋白质协同作用,维持细胞内铜水平的微妙平衡。然而,铜代谢稳态的破坏可能导致细胞内铜的过度积累,最终导致铜死亡。
用过量的铜和铜离子载体处理细胞会诱导细胞发生铜死亡,且依赖线粒体呼吸的癌细胞对铜死亡表现出更高的敏感性。FDX1是铜死亡发生所需的核心蛋白,其与LIAS的相互作用来实现的其功能。LIAS是催化硫辛酸生物合成最后一步的酶,并充当蛋白质脂酰化的关键上游调节因子。蛋白质硫辛酰化是一种翻译后修饰,这种修饰对于调节线粒体TCA循环的关键代谢酶的功能不可或缺,其中就包括丙酮酸脱氢酶复合物的重要亚基DLAT,研究发现LIAS和DLAT的基因缺失可使癌细胞对铜死亡产生抵抗。尽管蛋白质脂酰化引发铜死亡的确切机制仍不清楚,但已经观察到铜和硫辛酸化DLAT之间的直接结合促使硫辛酸化蛋白在铜死亡过程中发生寡聚化。这种现象提出了一种有趣的可能性,即所产生的蛋白质聚集体可能会引发细胞毒性并诱导铜死亡发生(图2)。
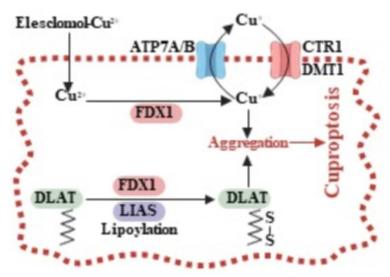 图2 铜死亡的关键调节因子
图2 铜死亡的关键调节因子鉴于铜死亡与线粒体代谢有着密切的联系,并且在依赖有氧呼吸的癌细胞中更容易诱导铜死亡发生,因此靶向铜死亡已成为潜在抗癌治疗干预的潜在途径。铜离子载体elesclomol已被证明能够选择性地将铜运送到线粒体引发细胞铜死亡,而癌细胞从糖酵解到氧化磷酸化的转变使它们对于elesclomol的作用更为敏感,值得注意的是临床观察结果与在癌细胞系中进行的研究相一致。Disulfiram作为一种铜结合化合物,它可通过在还原性细胞内环境中释放铜,从而增加细胞内的铜水平。最近的研究表明,双硫仑和铜的联合应用会引发肿瘤细胞铜死亡,并且在临床研究中双硫仑穿越血脑屏障的能力增强了其抗癌潜力。NSC319726是另一种用于癌症治疗的铜离子载体,其能够通过铜结合诱导癌细胞发生铜死亡。总的来说铜离子载体作为有前景的癌症治疗剂所具有的巨大潜力,未来的研究将致力于阐明铜死亡在协调这些铜离子载体的抗肿瘤功效中的作用,以及揭示利用铜死亡进行癌症治疗的其他策略。
双硫死亡
双硫死亡最近被认为是一种与细胞凋亡和铁死亡等其他死亡方式不同的调节性细胞死亡方式,其中SLC7A11是关键参与者。胱氨酸是一种溶解度极低的氨基酸,当它在细胞质中积累到高水平时,会对细胞产生显着的毒性。SLC7A11高表达的癌细胞将大量的胱氨酸转运到胞质中,需要将胞质胱氨酸快速转化为溶解度更高的半胱氨酸,这种转化需要 NADPH作为关键的还原剂,而胞质中的NADPH 被大量消耗。癌细胞高度依赖葡萄糖来提供必要的NADPH并维持胱氨酸向半胱氨酸的快速转化。当缺乏葡萄糖并因此缺乏 NADPH供应时,这些细胞会遭遇NADPH耗尽。这将导致细胞内胱氨酸和其他二硫键分子异常积聚,最终形成二硫键应激。二硫键应激触发肌动蛋白细胞骨架蛋白中的异常二硫键键合。这反过来又破坏了肌动蛋白网络的结构及其与质膜的分离,最终导致双硫死亡(图3)。
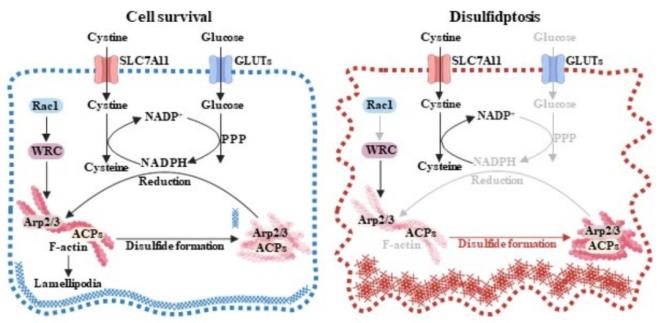 图3 双硫死亡的调控机制
图3 双硫死亡的调控机制双硫死亡的发现凸显了SLC7A11高癌细胞的代谢脆弱性,通过针对其对葡萄糖和 NADPH依赖,阐明了潜在的治疗途径。SLC7A11被证明可以抑制细胞凋亡和铁死亡,因此SLC7A11高的肿瘤可能对旨在诱导铁死亡和/或细胞凋亡的疗法表现出耐药性。然而,这些癌细胞对双硫死亡的易感性为SLC7A11高的恶性肿瘤干预治疗提供了新途径。临床前研究表明,与SLC7A11低水平的肿瘤相比,SLC7A11高水平的肿瘤对葡萄糖转运蛋白 (GLUT) 抑制剂表现出更高的敏感性。因此在临床前和临床环境中进一步探索和验证双硫死亡靶向策略可能会开创SLC7A11高癌症精准医学的新时代。
溶酶体锌死亡
在快速增殖的癌细胞中溶酶体功能通常会上调以满足其能量需求。TRPML1是一种对 Ca2+和 Zn2+具有双重渗透性的阳离子通道。Zn2+是正常细胞过程中不可缺少的微量元素,但胞内Zn2+的过量释放会阻碍线粒体功能并导致线粒体功能障碍、细胞能量耗尽并最终导致细胞死亡。特异性合成激动剂ML-SAs激活TRPML1促使溶酶体释放Zn2+,最终导致线粒体损伤和ATP迅速耗尽,最终导致细胞发生溶酶体锌死亡。重要的是,TRPML1高表达的细胞对于溶酶体锌死亡表现出更为明显的易感性。在小鼠黑色素瘤模型中,ML-SAs在抑制体内肿瘤进展方面表现显着,可以选择性地根除转移性黑色素瘤细胞,同时保持正常细胞不受伤害,因此靶向溶酶体锌死亡是潜在治疗黑色素瘤的有效策略(图4)。
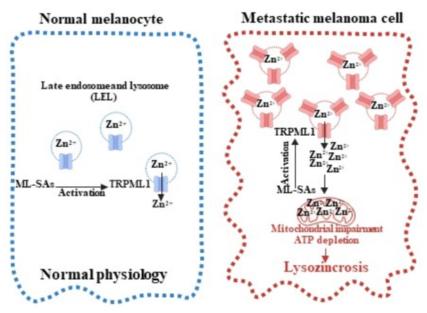 图4 溶酶体锌死亡的关键调节因子
图4 溶酶体锌死亡的关键调节因子碱死亡
细胞内pH平衡的破坏会导致酸化或碱化,最终导致细胞死亡。选择性抑制剂JTC801能够通过阻断OPRL1来触发细胞内碱化,从而诱导新型的细胞死亡形式碱死亡。从机制上讲,JTC801诱导碱死亡涉及NF-κB活性,阻断NF-κB通路可抑制碱死亡发生。细胞内 pH值的关键调节分子CA9参与了这一过程,NF-κB激活抑制CA9,促进碱死亡发生。最近的研究结果表明碱中毒是治疗多种癌症类型的一种有前景的方法。例如,JTC801在胰腺癌小鼠模型中显示出抑制肿瘤生长的功效,且无明显毒性。此外,JTC801诱导的碱中毒显示出抑制耐药急性髓性白血病细胞生长的潜力。碱死亡利用了癌细胞pH失调的弱点,有望成为一种新型治疗策略(图5)。
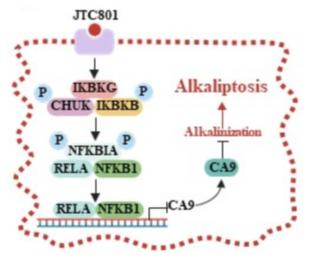 图5 碱死亡调节机制
图5 碱死亡调节机制总结
对癌症代谢性细胞死亡机制的探索揭示了其应用于肿瘤治疗的潜力。但它们也强调了一些需要进一步探索的重要问题。例如凋亡中的 BAX 和 BAK、坏死性凋亡中的MLKL以及细胞焦亡中的gasdermin D,在执行细胞自杀程序中发挥着关键作用。有趣的是,本文中讨论的细胞死亡途径的执行并不涉及此类成孔蛋白;相反,这些细胞死亡途径的共同特征是它们是由各种金属或营养素的消耗或超载导致的细胞代谢失衡诱导的,强调了细胞代谢与多种细胞死亡方式的调节之间复杂的相互作用。未来的研究可能会揭示这些细胞破坏途径之间更多的机制相似性和差异。
未来研究的另一个重要领域是这些细胞死亡机制之间的联系。如铁死亡和双硫死亡之间的联系以SLC7A11介导的胱氨酸转运为例,其中胱氨酸转运抑制铁死亡但促进双硫死亡。最近的研究揭示了铁死亡和铜死亡之间也存在着一定的联系。例如,GSH是GPX4介导的铁死亡防御的关键辅助因子,同时GSH也可以充当细胞内铜螯合剂抑制铜死亡发生。此外,最近的一项研究表明,铜通过诱导GPX4自噬降解来促进铁死亡,而铜螯合剂反过来又表现出抑制铁死亡的能力。这些研究共同强调了铁死亡、铜死亡和双硫死亡之间复杂的相互作用。预计进一步的研究将揭示代谢性细胞死亡之间的更多联系,对这些联系的全面理解可以为肿瘤的新型治疗干预措施提供新的见解(图6)。
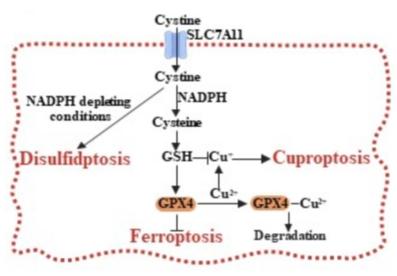 图6 铁死亡、双硫死亡和铜死亡之间的联系
图6 铁死亡、双硫死亡和铜死亡之间的联系从临床转化的角度来看,癌症治疗的一个巨大挑战在于实现选择性破坏癌细胞,同时保护正常细胞和免疫系统。尽管本综述中讨论的死亡诱导化合物已在癌细胞系和/或临床前模型中表现出功效,并且这些细胞死亡模式已揭示了特定癌症中的代谢敏感性,但将这些发现转化为对癌症患者有意义的治疗需要全面的临床前和临床实践。
毛超博士与王敏硕士为本文共同第一作者,甘波谊教授为本文通讯作者。
原文链接: https://academic.oup.com/proteincell/advance-article/doi/10.1093/procel/pwae003/7617526?login=true
本文转载自公众号“BioArt”
 中国生物物理学会官方订阅号,为BSC会员及生物物理领域专业人士服务。
中国生物物理学会官方订阅号,为BSC会员及生物物理领域专业人士服务。投稿及授权请联系:bscoffice@bsc.org.cn。
原标题:《【科技前沿】Protein & Cell 综述 | 甘波谊团队总结代谢性细胞死亡的机制以及癌症中的作用》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2026 上海东方报业有限公司

