- +1
【复材资讯】氟离子电池用“受体-多氟态”新概念电解液
01
研究背景
氟离子电池(FIBs)因其超高的理论能量密度、无枝晶的安全性和丰富的资源储备而受到了广泛关注。尽管已经提出了多种阴离子受体来解决无机氟盐在有机溶剂中的不溶性问题,但氟离子从受体中解离的困难导致氟离子电池的寿命短且比容量低。本文通过设计一种“受体-多氟态”,展示了一种具有超长寿命和超高比容量的氟离子电池。以三苯基氯化锑(TSbCl)作为电解质中的新型阴离子受体,其高路易斯酸性促进了氟化铯的完全解离,形成的TSbCl-F配合物可以进一步与氟离子相互作用,形成“受体-多氟态”。在电极-电解质界面释放氟离子时,“受体-多氟态”之间转化的能量显著低于TSbCl-F配合物直接解离所需的能量。该策略结合了氟盐的高解离能力和电极-电解质界面释放氟离子的低热力学障碍。这种电解质设计使得FIBs具有持久的可逆氟化/脱氟反应(超过3700次循环,库仑效率高达99.5%,电压极化仅为30 mV)和超高可逆容量(在100 mA/g下循环40次后容量为580 mAh/g)。此外,本工作还实现了高输出电压的FIBs(放电平台为2.9 V)和更大尺寸的软包电池(可逆容量为530 mAh/g)。
02
文章简介
近日,中国科学院上海硅酸盐研究所李驰麟研究员科研团队首次提出了一种“受体-多氟态”电解液体系。该电解液使用三苯基二氯化锑(简称TSbCl)作为受体,并使用氟化铯(CsF)作为氟盐。TSbCl由于Sb(Ⅴ)具有强路易斯酸性,被认为是一种能够有效解离氟化物盐的阴离子受体。当TSbCl与氟离子结合形成六配位锑化合物(TSbCl-F)后,TSbCl上的氯位点可以进一步被氟取代,形成多氟受体态(TSbCl-nF,n = 2,3)。TSbCl-nF与TSbCl-(n-1)F之间的转化能量显著低于经典的单氟受体态(acceptor-F)向受体态(acceptor)转化的能量,从而促进了氟离子从阴离子受体-氟离子复合物中的解离。极低的热力学界面阻力导致铜基充电产物以更高氧化态(+2.5)出现,从而提升了FIBs的比容量。多氟受体态的存在使得电解质能够在电极上轻松接受和释放大量氟离子,从而减少游离氟离子的存在以及游离氟离子与酸性原子之间的潜在副反应。这种效应因此维持了电极-电解质界面的长期稳定性。在铅基半电池(CuF2//Pb构型)中,含有CsF-TSbCl的二甲基亚砜(DMSO)电解质在0.1 mA/cm2的电流密度和0.1 mAh/ cm2的容量下,经过3700次循环后仍保持99.5%的平均库仑效率。相应的对称电池在相同电流密度下表现出超过1700小时的长寿命。CuF2//Sn+SnF2全电池在100 mA/g的电流密度下表现出580 mAh/g的可逆容量,并在100次循环后仍保持387 mAh/g的容量。高输出电压的CuF2//Li电池(放电平台为2.9 V)和CuF2//Sn+SnF2软包电池(可逆容量为530 mAh/g)也表现出优异的可逆性和长寿命。该工作为开发高比容量和长寿命的氟离子电池提供了一种新概念的阴离子受体电解质设计。
 03
03本文要点
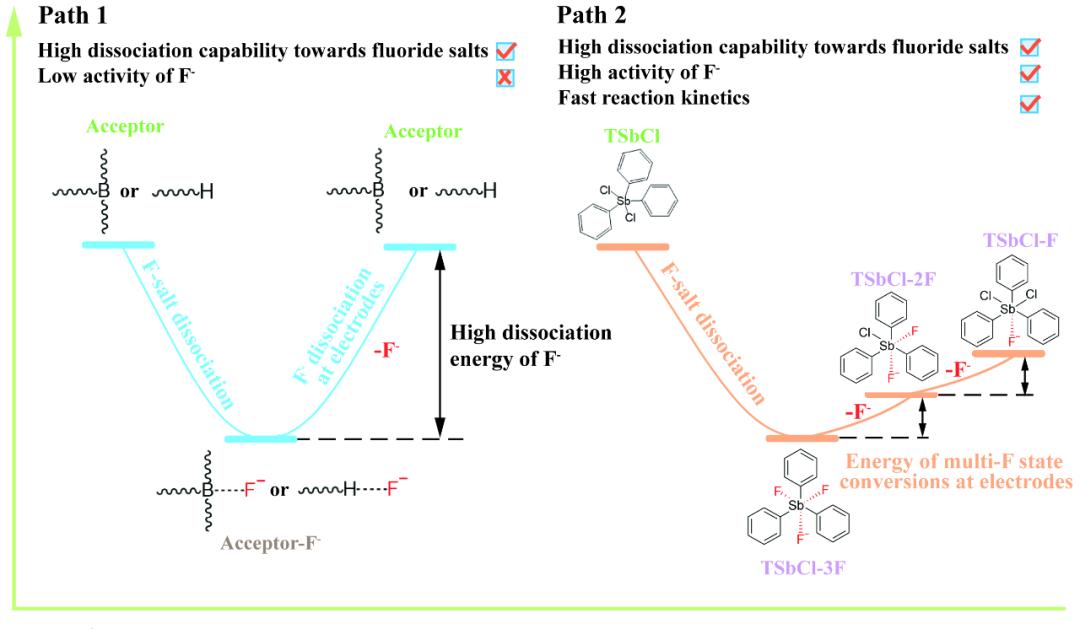
要点一:“受体-多氟态”电解液设计
DFT计算显示,TSbCl对氟离子的吸附能为-3.54 eV,显著低于氟离子取代TSbCl分子上的氯所需的反应能(-1.80 eV)。这一比较说明,TSbCl优先形成六配位的锑化合物(TSbCl-F)。如果在电解质配方中TSbCl与氟离子的化学计量比小于1:1,则过量的氟离子可以进一步与六配位锑化合物中的氯交换,导致“受体-多氟态”的形成。这些“受体-多氟态”之间转变的能量(例如,TSbCl-F与TSbCl-2F之间为-1.53 eV,TSbCl-2F与TSbCl-3F之间为-1.62 eV)远低于TSbCl-F分子分解为TSbCl和氟离子的解离能,从而促进了电极处的阴离子受体-氟离子复合物的氟离子解离。在19F NMR光谱中,观察到了分别归属于TSbCl-F、TSbCl-2F和TSbCl-3F的三个峰。XRD确认电解质中的沉淀物为高结晶度的CsCl。这些结果表明CsF完全解离,存在“受体-多氟态”,且在CsF-TSbCl-DMSO电解质中不存在自由的氯离子。制备电解质所涉及的反应如下:
TSbCl + CsF → TSbCl-F + TSbCl-2F + TSbCl-3F + Cs+ + CsCl(沉淀)
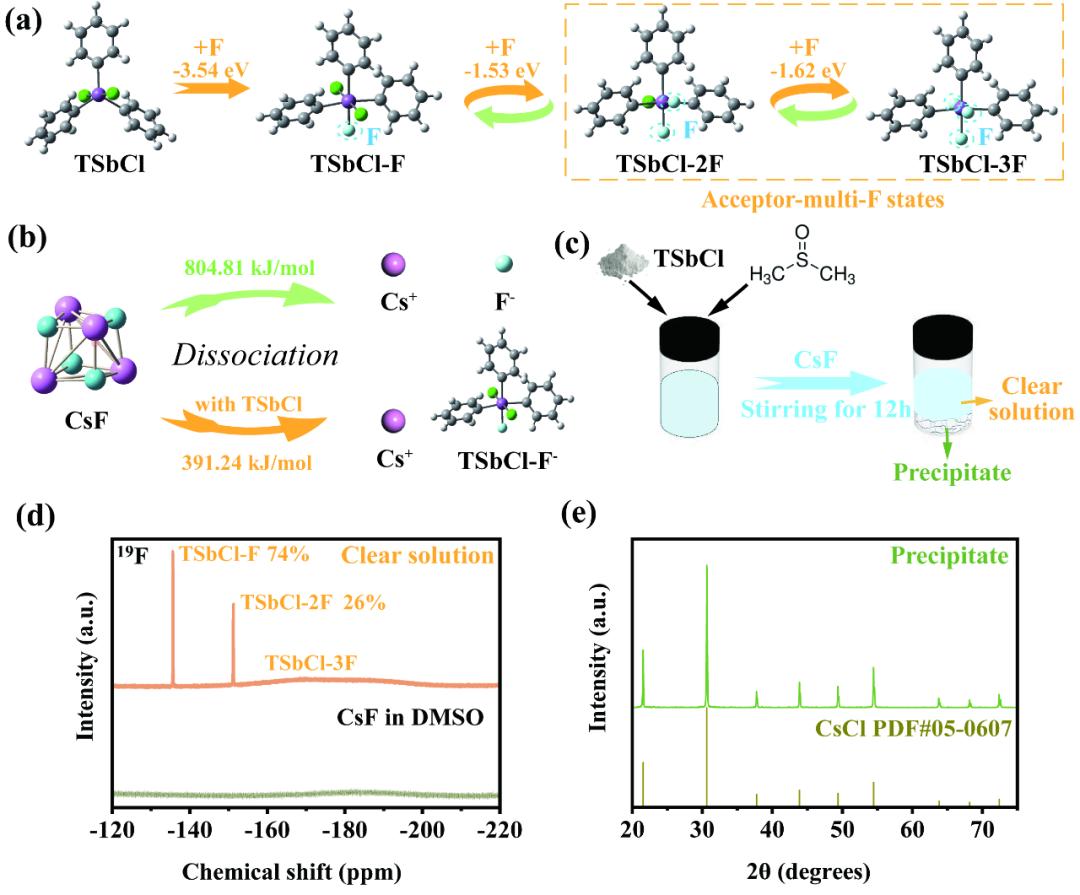 图1 “受体-多氟态“电解液设计
图1 “受体-多氟态“电解液设计要点二:“受体-多氟态”电解液的半电池和对称电池性能
组装的半电池在0.1 mA/cm2的电流密度下,表现出超过3700小时的稳定循环,库仑效率约为99.5%,在整个循环过程中电压曲线波动极小。氟化与脱氟之间的极化保持在30 mV以内,无明显过电位。即使与成熟的锂金属电池系统相比,这种循环性能也尤为突出。分别以Mg+MgF2、Sn+SnF2和Pb+PbF2为对称电池的电极,在0.1 mA/cm2和0.1 mAh/cm2的条件下,Mg+MgF2//Mg+MgF2电池的循环寿命超过1700小时。然而,MgF2的高晶格能导致氟化/脱氟过程中的极化高达150 mV。Pb+PbF2//Pb+PbF2对称电池的极化较小,仅为30 mV。但在230小时后,界面退化和铅溶解导致极化显著增加。相比之下,Sn+SnF2//Sn+SnF2对称电池不仅实现了超过1700小时的稳定循环,且极化低至25 mV。Sn+SnF2电极的对称电池随着电流密度的增加,当电流密度高达2 mA/cm2时,过电位从25 mV升至420 mV,且每个阶段的过电位均高度稳定,波动极小。当电流密度回到0.1 mA/cm2时,过电位可恢复至25 mV。与现有关于氟离子电池(FIBs)的报道相比,“受体-多氟态”电解质实现了电流密度提升20倍、过电位显著降低以及循环寿命延长4-5倍。
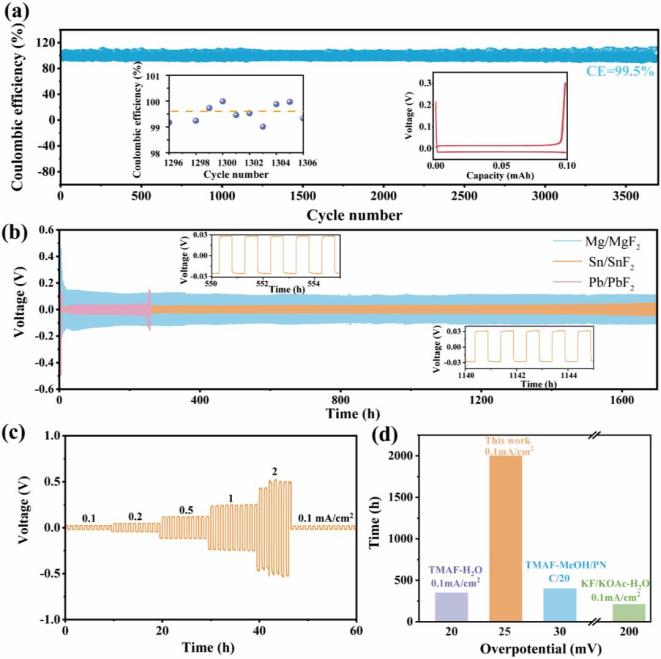 图2 “受体-多氟态”电解液的半电池和对称电池性能
图2 “受体-多氟态”电解液的半电池和对称电池性能要点三:“受体-多氟态”电解液的全电池性能
组装的全电池在100 mA/g的电流密度下,初始放电比容量高达772 mAh/g,放电平台在-0.03 V vs. Sn2+/Sn(即2.87 V vs. Li+/Li)。在最初的40个循环中,比容量保持在580 mAh/g,经过100次循环后,该电池的比容量保持在387 mAh/g,且在整个循环过程中库仑效率始终保持在100%。CuF2//Sn+SnF2全电池的极化较小,仅为0.06 V。组装的氟离子软包电池在20 mA/g的电流密度下,其初始放电比容量为530 mAh/g,放电平台在-0.01 V vs. Sn2+/Sn(即2.89 V vs. Li+/Li)。经过50次循环后,该软包电池仍保持200 mAh/g的可逆容量,且在这些循环中库仑效率约为98%。与目前报道的使用液体电解质的FIBs相比,我们利用“受体-多氟态”电解质的全电池表现出最低的极化、超长的寿命、最高的比容量以及在高电流密度下优异的容量保持率。组装的CuF2//Li电池在首次循环中表现出580 mAh/g的放电比容量,并有两个放电平台,分别约为2.5 V和2.3 V。
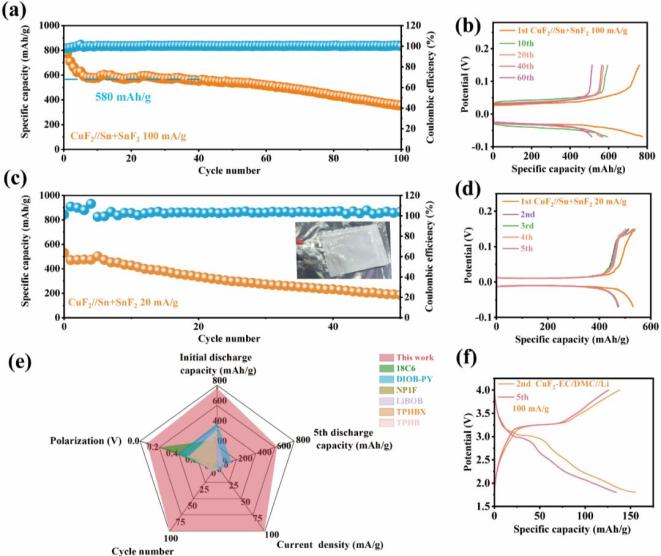 图3 “受体-多氟态”电解液的全电池性能
图3 “受体-多氟态”电解液的全电池性能要点四:“受体-多氟态”电解液中的反应机制
放电后,正极由大量纳米级颗粒组成。SAED显示,主要的放电产物是Cu20Sn6。少量Sn的存在是由于部分Sn从负极溶解到电解质中,而Cu与Sn的合金化有利于阻止Cu从正极溶解。HRTEM进一步显示出与Cu20Sn6的(200)晶面对应的清晰晶格条纹,证实了Cu20Sn6为主要放电产物。在充电后,Cu-Sn纳米颗粒的边界在再氟化过程中扩展,相应的界面合并形成更大的微米级颗粒。SAED显示,主要的充电产物是CsCu2F6。HRTEM进一步证实,充电产物由CsCu2F6和CuF2组成。CsCu2F6区域位于靠近正极-电解质界面(CEI)层的外侧,而CuF2主要存在于远离CEI的内层。这一结果表明,外部的Cu在氟化过程中倾向于与CEI内的Cs+结合,其中较大尺寸的Cs+可以稳定晶格中的高价Cu2.5+。我们采用DFT计算来进一步验证CsCu2F6生成的可行性并了解Cu的氟化路径。由CuF2与Cs+反应形成CsCu2F6的反应能为-11.93 eV。尽管CuF2的进一步氟化在热力学上是有利的,但由于Cs+的扩散动力学阻碍,该反应倾向于在CEI附近发生。Sn+SnF2电极的XRD结果显示,放电后,Sn的特征峰几乎消失,而SnF2的峰增强,表明Sn几乎完全氟化。在重新充电后,SnF2的峰显著减弱,而Sn的峰重新出现,表明SnF2脱氟为Sn。
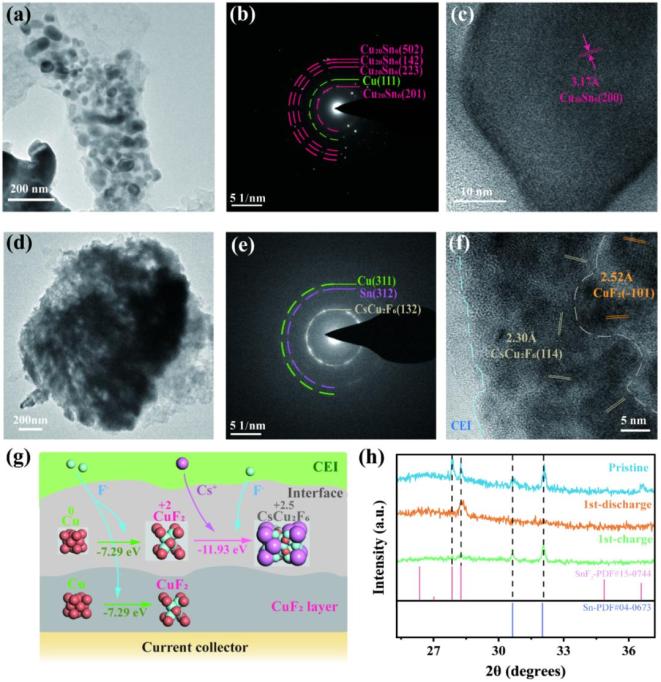 图4 “受体-多氟态”电解液中的反应机制
图4 “受体-多氟态”电解液中的反应机制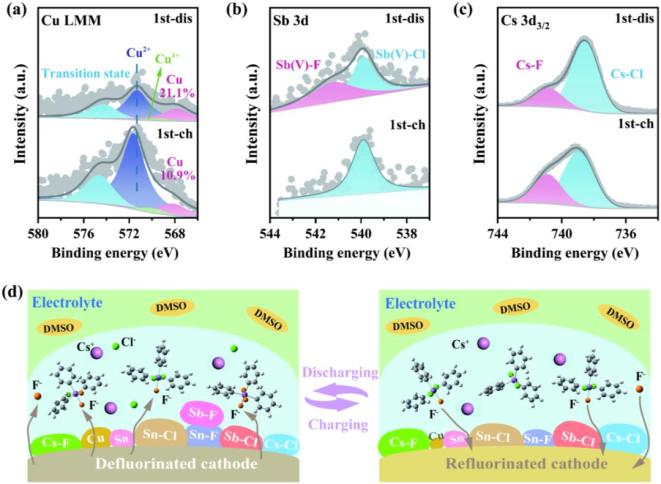 图5 “受体-多氟态”电解液中的正极在循环过程的表面组分演化
图5 “受体-多氟态”电解液中的正极在循环过程的表面组分演化原文链接:https://advanced.onlinelibrary.wiley.com/doi/10.1002/adma.202415106
免责声明:中国复合材料学会微信公众号发布的文章,仅用于复合材料专业知识和市场资讯的交流与分享,不用于任何商业目的。任何个人或组织若对文章版权或其内容的真实性、准确性存有疑议,请第一时间联系我们。我们将及时进行处理。
继续滑动看下一个轻触阅读原文
 中国复合材料学会向上滑动看下一个
中国复合材料学会向上滑动看下一个原标题:《【复材资讯】氟离子电池用“受体-多氟态”新概念电解液》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2026 上海东方报业有限公司

