- +1
中国完成全球首个新冠随机对照药物临床试验,评价“克力芝”
新冠肺炎COVID-19的全球第一项随机对照临床试验结果公布。北京时间3月19日,国家呼吸疾病临床研究中心、中日友好医院、武汉市金银潭医院、北京地坛医院、北京协和医学院等团队的研究人员联合在顶级医学期刊《新英格兰医学杂志》(NEJM)在线发表了研究论文“洛匹那韦-利托那韦(商品名‘克力芝’)治疗重症COVID-19成人住院患者的试验”。
该项研究的通讯作者为中日友好医院副院长、呼吸与危重症医学科主任曹彬,武汉市金银潭医院院长张定宇,中国工程院副院长、中国医学科学院北京协和医院院长、国家呼吸疾病临床医学研究中心主任王辰。

值得注意的是,这是新冠疫情暴发以来,世界顶级医学杂志首次发表治疗COVID-19的临床试验结果,也是在近20年新发传染病疫情期间发表的屈指可数的药物临床试验结果。
这项研究的总体结论为:在重症COVID-19成人住院患者中,与标准治疗相比,研究团队未观察到洛匹那韦-利托那韦治疗有益。
研究团队在讨论环节提到,这项随机试验发现,对COVID-19重症患者采用洛匹那韦–利托那韦治疗加标准支持治疗,与仅提供标准支持治疗相比,重症患者的临床改善或死亡率降低并不明显。但是,在改良意向性治疗分析中,研究排除了3名早期死亡的患者之后,两组之间的临床改善中位时间显示出了差异,尽管幅度不大,但是有统计学显著性,具体来说中位数是15天vs.16天。
该研究发现:接受洛匹那韦-利托那韦治疗的患者出现严重并发症(急性肾损伤和继发感染)或需要无创或有创机械通气以治疗呼吸衰竭的数量少于未接受治疗的。根据这些观察结果,研究团队表示,需要进一步的研究以确定在特定疾病阶段接受洛匹那韦–利托那韦治疗,是否可以减少COVID-19的某些并发症。
值得注意的是,该试验的总体死亡率是22.1%,仍大大高于COVID-19住院患者的初步描述性研究报告中11%至14.5%的死亡率,这表明纳入试验的是重病患者。
洛匹那韦利托那韦是两种蛋白酶抑制剂洛匹那韦和利托那韦构成的合剂,商品名为克力芝。2000年被美国FDA批准上市用于艾滋病的抗病毒治疗,虽然服用药物会带来腹泻、呕吐、血脂高等副作用,但由于抗病毒效果好、耐药屏障高,目前仍作为主要的抗HIV治疗药物应用于临床。值得一提的是,洛匹那韦利托那韦也是此次疫情暴发之后最早被使用的抗病毒老药之一。
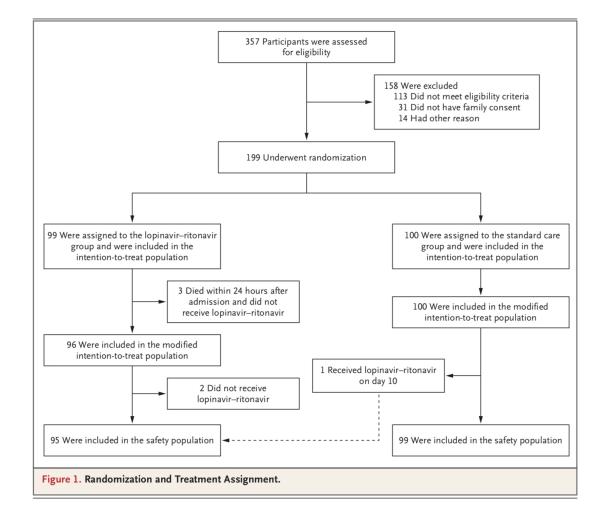
该临床试验于2020年1月9日通过伦理审查,1月18日至2020年2月3日(最后一例患者的入组日期)在武汉市金银潭医院开展。共计199例实验室确诊且符合入组条件的新冠病毒感染患者被随机分组。其中100例被分配至标准治疗组;99例被分配至洛匹那韦-利托那韦组,也就是说在标准治疗的基础上,增加洛匹那韦-利托那韦治疗(各400mg、100mg,每日两次,疗程14天)。
值得注意的是,有5例被分配到洛匹那韦-利托那韦组的患者并未接受洛匹那韦-利托那韦治疗(其中3人在24小时内死亡),但为了最大限度地保留随机化的信息,同样被纳入了意向治疗(ITT)分析。研究团队同样还进行了一项排除3例早期死亡患者的改良意向性治疗(mITT)分析。
但该项研究最终选取ITT分析集作为主要终点结果。这项研究的主要研究者在公众号“NEJM医学前沿” 撰写的一份述评提到:每个科研攻关团队都希望本团队提出的干预措施和药物显示特效,我们作为该项目的研究者,也有着强烈的愿望,希望该药物能够有效。但是本着研究结果要经得起质疑和检验的原则,NEJM杂志编辑和我们都采取了谨慎的态度选取ITT分析集作为主要终点结果,为本文下主要结论,而并未选取mITT分析集的主要终点结果。
在ITT分析集中,洛匹那韦-利托那韦组中位改善时间16天,14天改善率45.5%,最终28天累计改善率78.8%;标准治疗组,中位改善时间16天,14天改善率30%,最终28天累计改善率70%。两组改善的风险比(HR)是 1.31(95% CI,0.95-1.80),P=0.09。
在mITT分析集中,洛匹那韦/利托那韦组中位改善时间15天,14天改善率46.9%,最终28天累计改善率81.3%;标准治疗组,中位改善时间16天,14天改善率30%,最终28天累计改善率70%。两组改善的风险比(HR)是 1.39(95% CI,1.00-1.91),P=0.0377。
此外,在亚组分析中,研究团队发现患者发病12天内使用洛匹那韦/利托那韦,获益趋势更为明显。
199例患者纳入临床试验
从2019年12月开始, 新型冠状病毒(SARS-CoV-2)在全球引发了称为COVID-19的呼吸系统疾病暴发。COVID-19的完整疾病谱范围是从轻度自限性呼吸道疾病到重度进行性肺炎、多器官衰竭和死亡。迄今为止尚无针对冠状病毒感染的特异性治疗药物。
此前的研究显示,2003年严重急性呼吸系统综合征(SARS)出现后,对已获批准的药物进行筛选后发现,洛匹那韦(人类免疫缺陷病毒[HIV]1型的天冬氨酸蛋白酶抑制剂)对SARS-CoV病毒具有体外抑制活性。
论文提到,利托那韦与洛匹那韦联用,主要是通过抑制细胞色素P450来延长洛匹那韦的血浆半衰期。2004年发表的一项开放标签研究提示,与仅接受利巴韦林治疗的历史对照组相比,利巴韦林加用洛匹那韦-利托那韦(分别为400 mg和100 mg)降低了SARS患者的不良临床结局(急性呼吸窘迫综合征ARDS或死亡)风险以及病毒载量。
然而,由于上述研究未进行随机化,也没有设置同期对照组,并且联用了糖皮质激素和利巴韦林,因此难以评估洛匹那韦-利托那韦的作用。
类似地,在体外试验和动物模型中,洛匹那韦对中东呼吸系统综合征冠状病毒(MERS-CoV)均有活性。并且有病例报告指出,洛匹那韦-利托那韦与利巴韦林和干扰素(IFN)α联用可清除病毒,使患者存活。
然而,目前尚无确实数据证明将该疗法用于人体的疗效。治疗MERS的一项临床试验(联用重组干扰素β-1b)目前正在进行中(在ClinicalTrials.gov注册号为NCT02845843)。
为了评估口服洛匹那韦-利托那韦治疗新冠病毒感染的疗效和安全性,研究团队在COVID-19成人住院患者中开展了一项随机、对照、开放标签试验LOTUS China(Lopinavir Trial for Suppression of SARS-Cov-2 in China)。
本试验的纳入标准如下:年龄≥18岁,诊断标本的RT-PCR结果呈阳性,胸部影像学检查确诊肺炎,呼吸周围空气时氧饱和度(SaO2)≤94%或氧分压(PaO2)与吸入氧浓度(FiO2)的比值(PaO2:FiO)≤300 mm Hg的男性和非妊娠期女性患者。
该项临床研究2020年1月18日至2020年2月3日(最后一例患者的入组日期)在武汉市金银潭医院开展。
研究团队以1:1的比例将符合参与试验标准的患者随机分成两组,分别接受为期14日的标准治疗联合每日两次洛匹那韦-利托那韦(400 mg和100 mg,口服)治疗、单独接受标准治疗。
根据患者的需要,标准治疗包括吸氧、无创和有创通气、抗生素、血管加压药、肾脏替代疗法和体外膜氧合(ECMO)。
作为呼吸衰竭严重程度的指标,为了平衡两组之间氧气支持的分布情况,研究团队根据患者入组时的呼吸支持方法对随机分组进行了分层:无氧气支持或者采用鼻导管或面罩的氧气支持,或者高流量氧无创通气,或者包括ECMO在内的有创通气。
在第0至至第28日、患者出院或患者死亡期间,由护士每日两次根据日记卡评估患者状况,日记卡中记录了7分等级量表数据和安全性数据。
研究团队设置的主要终点是至临床状况改善的时间,其定义为从随机分组至7分等级量表改善2分(与随机分组时的状况相比)或至出院的时间,以先发生的一项为准。
等级量表在之前已被用作重症流感住院患者临床试验的终点。7分等级量表包括以下等级:1.未住院,且可继续从事日常活动;2.未住院,但无法继续从事日常活动;3.住院治疗,不需要吸氧;4.住院治疗,需要吸氧;5.住院治疗,需要经鼻高流量氧疗、无创机械通气或这两者;6.住院治疗,需要ECMO、有创机械通气或这两者均需要;7.死亡。
其他临床结局包括在第7日和第14日时采用7分等级量表评估的临床状况、28日死亡率、机械通气持续时间、生存者的住院时长,以及从治疗开始至死亡的时间(天数)。病毒学指标包括随时间推移,检出病毒RNA的患者比例,以及病毒RNA滴度曲线下面积(AUC)测定值。
安全性结局包括治疗期间发生的不良事件、严重不良事件和提前停止治疗。
研究团队在第1日(服用洛匹那韦-利托那韦之前)及第5、10、14、21和28日(直至患者出院或死亡)采集了患者的口咽拭子样本,并进行了实时RT-PCR检测。并未因某一时间点的拭子检测结果呈阴性而停止之后的样本采集。
多项数据显示两组无明显差异,胃肠道不良事件发生率较高
在被随机分组的199例患者中,99例患者接受洛匹那韦-利托那韦治疗,100例患者接受单独标准治疗。在洛匹那韦-利托那韦组的99例患者中,94例(94.9%)接受了分配的治疗。
洛匹那韦-利托那韦组有5例患者未接受洛匹那韦-利托那韦治疗,其中3例的原因是在随机分组后24小时内早期死亡,另外2例的原因是随机分组后,主治医师拒绝开出洛匹那韦-利托那韦。
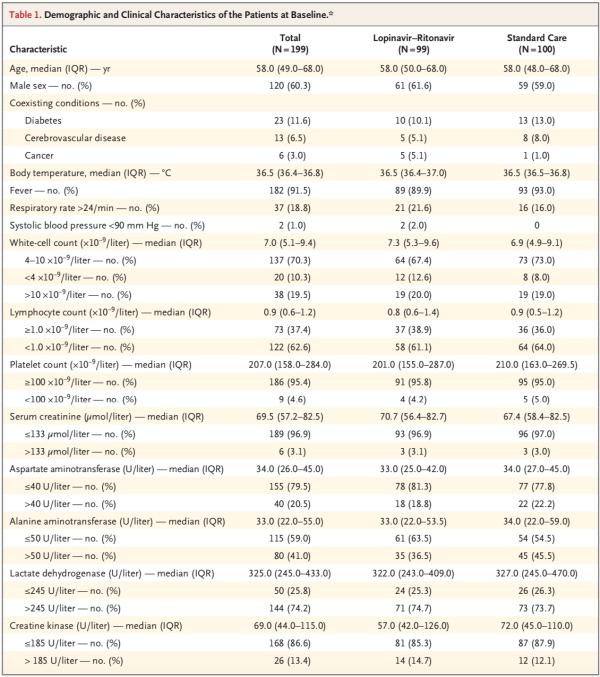
患者中位年龄为58岁(四分位距,49-68岁),男性患者占60.3%。出现症状和随机分组的中位间隔时间为13天(四分位距,11-16天)。
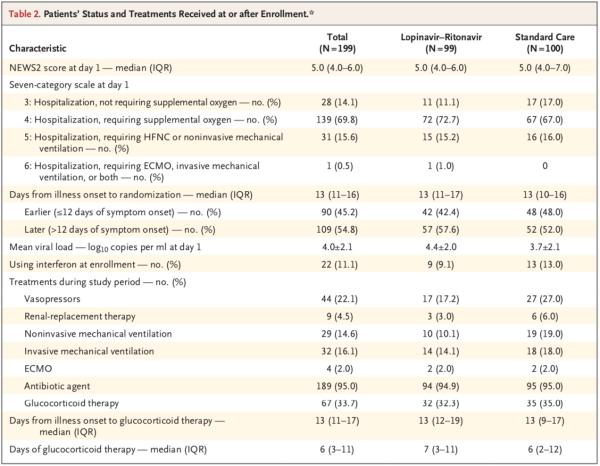
在人口统计学特征、基线实验室检查结果、等级量表评分分布情况或入组时的NEWS2评分方面,两组间无重要差异。在该试验期间,洛匹那韦-利托那韦组和标准治疗组接受全身糖皮质激素治疗患者的比例分别为33.0%、35.7%。
研究团队得出,在意向性治疗人群中,洛匹那韦-利托那韦组和标准治疗组患者至临床状况改善的时间无差异(中位数,16天vs. 16天;临床改善的风险比,1.31;95%置信区间,0.95-1.85;P=0.09)。
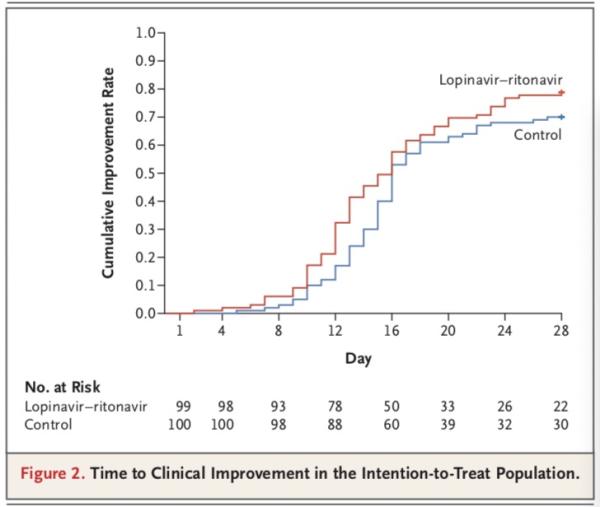
不过,在改良意向性治疗人群中,洛匹那韦-利托那韦组和标准治疗组患者至临床状况改善的中位时间分别为15天和16天(风险比,1.39;95%置信区间,1.00-1.91)。
在意向治疗人群中,症状出现后12日内接受洛匹那韦-利托那韦治疗与较早达到临床状况改善相关(风险比,1.25;95%置信区间,1.77-2.05),而较晚接受洛匹那韦-利托那韦治疗则与较早达到临床状况改善不相关(风险比,1.30;95%置信区间,0.84-1.99)。
研究团队在讨论环节提到,COVID-19患者在早期旧使用洛匹那韦–利托那韦治疗是否可能具有临床益处,这个问题很重要,也需要进一步研究。该研究发现与新冠病毒性肺炎在疾病的第二周内进展相符,并且与先前在SARS和严重流感中进行的抗病毒研究中观察到的治疗时间效应也相符。
在意向治疗人群中,如果根据入组时的NEWS2评分对至临床状况改善的时间进行评估,未观察到显著差异。此外,两组之间至临床状况恶化(定义为7分量表评分加重1分)的时间也无差异(临床改善的风险比,1.01;95%置信区间,0.76-1.34)。
次要结局方面,在意向性治疗人群(19.2% vs. 25.0%;差异,-5.8个百分点;95%置信区间,-17.3-5.7)或改良意向性治疗人群(16.7% vs. 25.0%;差异,-8.3个百分点;95% CI,-19.6-3)中,洛匹那韦-利托那韦组的28日死亡率在数值上均低于标准治疗组。
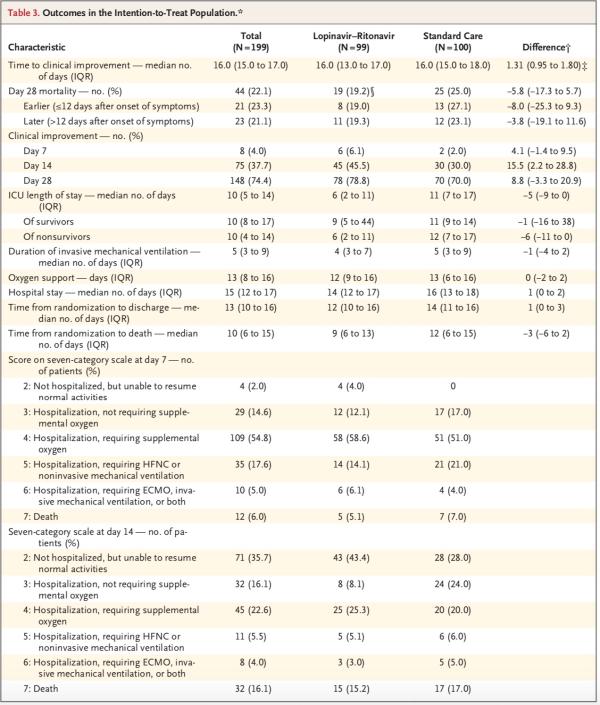
洛匹那韦-利托那韦组患者的重症监护病房(ICU)住院时间比标准治疗组患者短(中位数,6天vs. 11天;差异,-5天;95% CI,-9-0),并且从随机分组至出院的时间比标准治疗组短(中位数,12天vs. 14天;差异,1天;95% CI,0-3)。此外,第14日时,洛匹那韦-利托那韦组中临床状况改善的患者百分比高于常规治疗组(45.5% vs. 30.0%;差异,15.5个百分点;95% CI,2.2-28.8)。
其他结局(例如吸氧持续时间、住院时长和从随机分组至死亡的时间)无显著差异。
病毒学方面,随机分组时,洛匹那韦-利托那韦组患者咽拭子样本(获取知情同意后采集的样本)的基线病毒RNA载量平均值(±SD)略高于常规治疗组(4.4±2.0 log10 拷贝/毫升 vs. 3.7±2.1 log10拷贝/毫升)。
随着时间的推移,洛匹那韦-利托那韦组患者和标准治疗组患者的病毒RNA载量无差异,包括根据患病时间进行的分析)。
在任何采样时间上,洛匹那韦-利托那韦组和标准治疗组可检出新冠病毒RNA的患者百分比均相似(第5日,34.5% vs. 32.9%;第10日,50.0% vs. 48.6%;第14日,55.2% vs. 57.1%;第21日,58.6% vs. 58.6%;第28日,60.3% vs. 58.6%)。
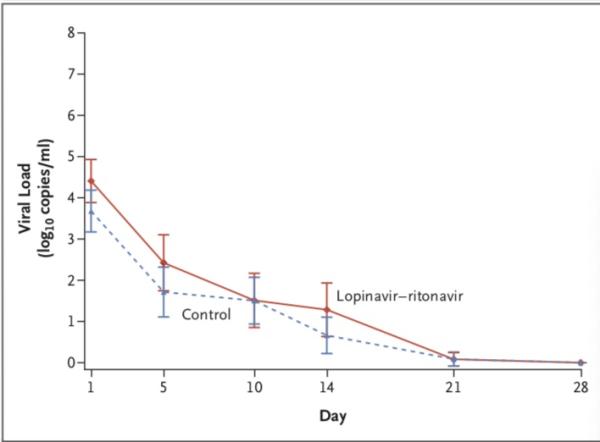
研究团队认为,与单独的标准支持治疗相比,未发现加入洛匹那韦–利托那韦治疗可减少病毒RNA载量,也未发现患者病毒RNA可检测时间的缩短。在试验结束时(第28天),仍在洛匹那韦–利托那韦治疗组的40.7%的患者中仍检测到新冠病毒的RNA。最近的一份报告显示,在严重疾病患者中,COVID-19的排毒时间中位时间为20天,有的可能长达37天。
该研究或当前研究均未发现洛匹那韦-利托那韦发挥重要抗病毒作用的证据。洛匹那韦–利托那韦对新冠病毒肺炎患者的治疗缺乏抗病毒作用的原因尚不确定,“但当前试验中使用的采样方法很可能也不是最佳的选择。”
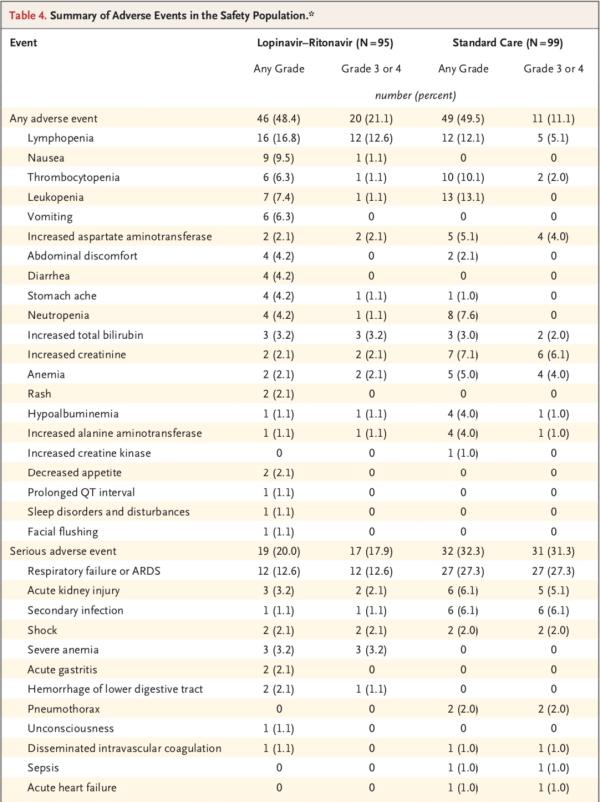
安全性研究显示,随机分组至第28日期间,洛匹那韦-利托那韦组和标准治疗组分别有46例患者(48.4%)、49例患者(49.5%)报告了不良事件。洛匹那韦-利托那韦组中胃肠道不良事件(包括恶心、呕吐和腹泻)的发生率高于常规治疗组。两组中有实验室检查结果异常的患者百分比相似。
共计51例患者发生了严重不良事件:洛匹那韦-利托那韦组19例和标准治疗组32例(表4)。洛匹那韦-利托那韦组发生了4起胃肠道严重不良事件,而标准治疗组未发生胃肠道严重不良事件。研究团队判定这4起事件均与试验药物相关。而标准治疗组患者的呼吸衰竭、急性肾损伤和继发感染发生率高于洛匹那韦-利托那韦组。
研究团队提到,在本试验中观察到的副作用引起了人们对使用更高或更长的洛匹那韦-利托那韦剂量方案以改善结局的担忧。
研究团队还判定,观察期间的所有死亡均与干预无关。
总体来说,研究团队发现对于严重的COVID-19患者,洛匹那韦–利托那韦治疗并不能显著加速患者的临床改善、降低死亡率或降低咽喉病毒RNA的可检测性。这些早期数据能为将来的研究提供参考,以评估该药物和其他药物治疗新冠病毒感染的可能性。
新发传染病特效药的窘境:有效治疗措施的探索时间窗极为狭窄
值得注意的是,该项研究的主要研究在前述述评文章中还披露了该项临床试验背后的一些信息。
首先,为什么选择洛匹那韦-利托那韦做临床试验,以及为什么选择在金银潭医院开展?
张定宇此前曾有所透露,“我们有一个先天的优势,因为洛匹那韦/利托那韦是抗艾滋药,我们医院是管艾滋病的,全省的艾滋病药全部在我们这。当时想着一个病人按14天来算,大概是需要56颗药,一瓶药120粒,可以给两个病人吃。按照这个算法我们大概有1000人份的药。所以我们很快在临床展开了,鼓励一些科室主任,如果有重病人的话,赶快给这个药,说不定有用。”
主要研究者在上述述评中写道:在疫情初期,面对部分重症患者病情快速恶化和难以逆转的难题,我们一直在思考有什么药物能够救治更多重症患者?
“其实我们对提出洛匹那韦/利托那韦的建议是很忐忑的,害怕医生不理解,甚至产生了退而求其次做队列观察的念头。但是,张定宇院长和他的团队给了中日医院团队坚定的支持。最后中日医院和金银潭医院两个团队合作共同完成COVID-19的全球第一项随机对照临床试验。”
他们同时提到,瑞德西韦是列为最重要评价的药物,因为该药体外抗病毒活性最强,动物实验验证有效,有I/II/III临床试验数据。“但由于瑞德西韦全球未上市,临床可及性不足,因此我们把瑞德西韦列为第二需要评价的药物。”
然而,该临床试验1月9日完成立项,但直到1月18日正式随机入组第一例患者,期间曲折颇多。
其中之一包括临床试验的注册。研究团队希望尽快在相应的临床试验平台上注册、公开研究信息,让同行监督保证临床试验公开透明。但ClinicalTrials.gov注册网站以“COVID-19是地方性疾病”拒绝我们注册,后改在中国临床试验注册平台上登记注册成功,但是因此而耽误了宝贵的时间。当他们完成该临床试验注册时,已经在入组第1例患者之后。
述评中写道,“这真是一个莫大的讽刺,1月初的时候,ClinicalTrials.gov注册网站怎么也没有想到短短两个月后COVID-19已经席卷全球。”
并且,在中日友好医院团队1月9日返回北京参加其他疫情防控后的1周时间里,随机系统中随机人数一直都是0。多次沟通后发现项目进展困难,“张定宇院长团队先说服临床医生在少数患者中尝试了使用,并发现吸入干扰素的操作会增加临床执行的困难,增加气溶胶暴露的风险。”
最后,研究团队对方案进行了修改,改用洛匹那韦-利托那韦单用作为干预措施。
中日友好医院团队1月23日再次回到金银潭医院。“我们不禁感慨万千,武汉当时疫情形式的严峻超过了我的想象。”
研究者们提到,面对新发突发呼吸道传染病,快速明确有效治疗药物十分困难。在尝试治疗SARS 和MERS的历史中,因诸多原因两次次错失证明洛匹那韦-利托那韦疗效和安全性的机会。
“连续错失两次探索治疗冠状病毒药物的机会,间接导致此次COVID-19全球流行无确切抗病毒药物的窘境。”研究团队认为,面对新发突发呼吸道传染病,有效治疗措施的探索时间窗极为狭窄,需要集中最宝贵的资源优先评价几种具有充分潜力的药物。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2026 上海东方报业有限公司

